

Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm
- PMID: 26352475
- PMCID: PMC4857894
- DOI: 10.1038/nature15365
Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm
Abstract
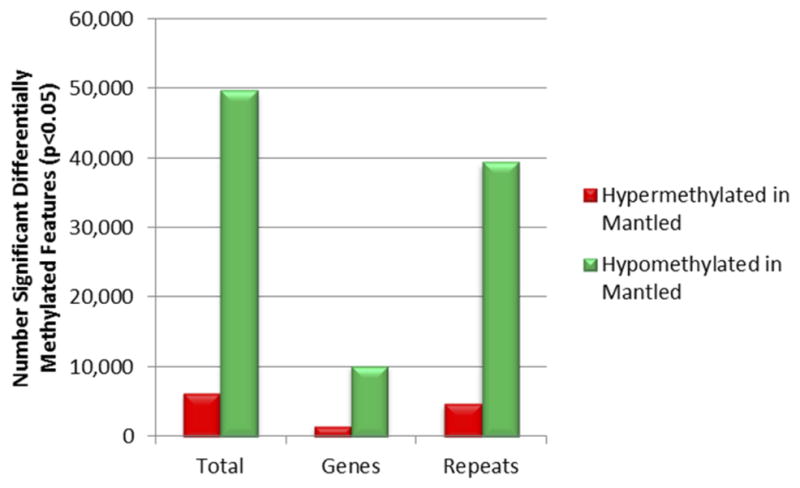
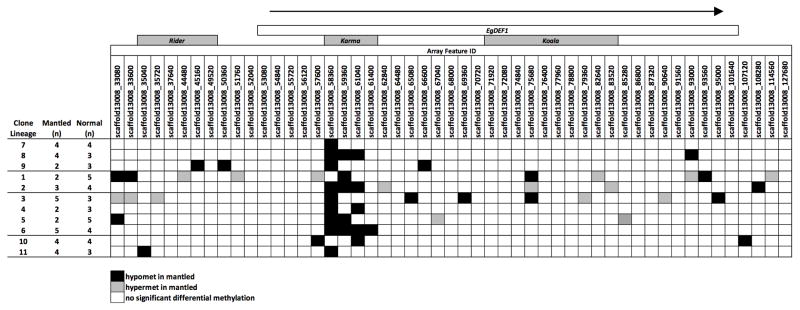
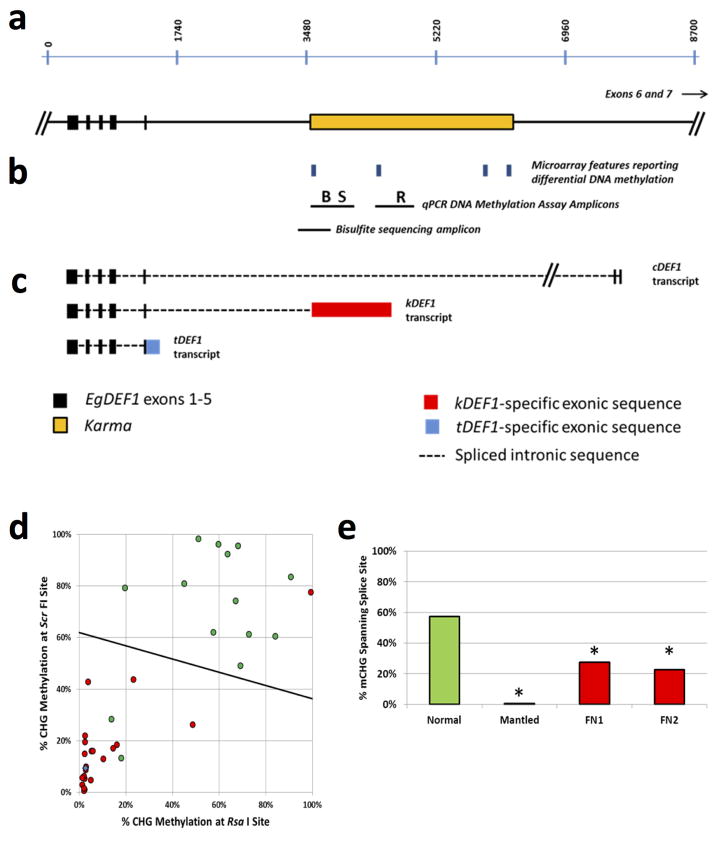
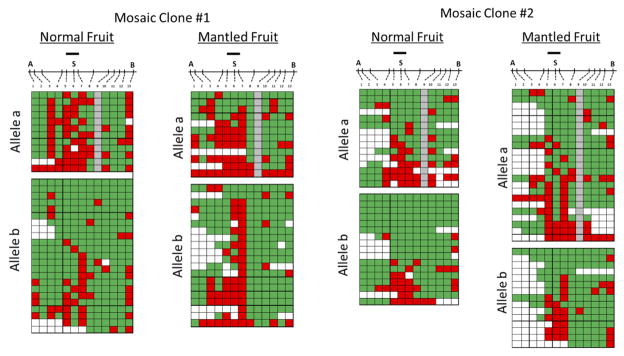
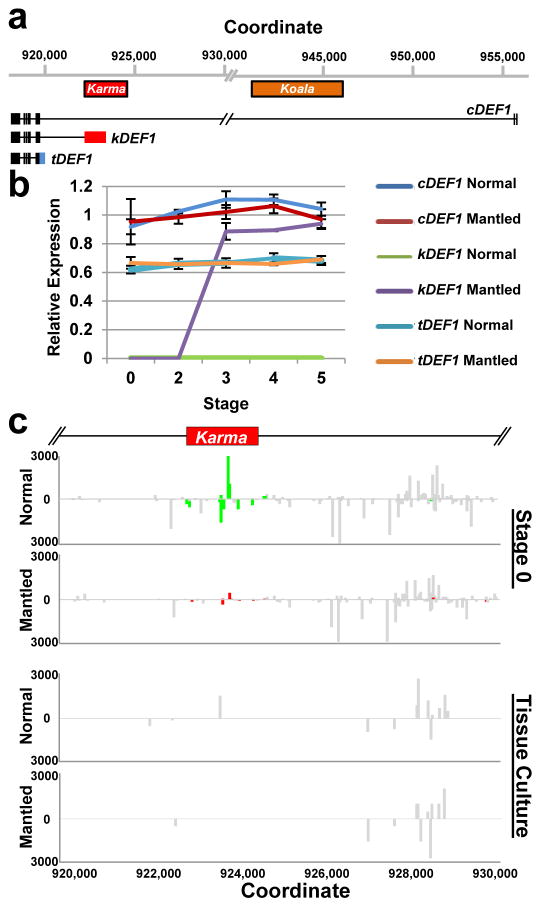
Somaclonal variation arises in plants and animals when differentiated somatic cells are induced into a pluripotent state, but the resulting clones differ from each other and from their parents. In agriculture, somaclonal variation has hindered the micropropagation of elite hybrids and genetically modified crops, but the mechanism responsible remains unknown. The oil palm fruit 'mantled' abnormality is a somaclonal variant arising from tissue culture that drastically reduces yield, and has largely halted efforts to clone elite hybrids for oil production. Widely regarded as an epigenetic phenomenon, 'mantling' has defied explanation, but here we identify the MANTLED locus using epigenome-wide association studies of the African oil palm Elaeis guineensis. DNA hypomethylation of a LINE retrotransposon related to rice Karma, in the intron of the homeotic gene DEFICIENS, is common to all mantled clones and is associated with alternative splicing and premature termination. Dense methylation near the Karma splice site (termed the Good Karma epiallele) predicts normal fruit set, whereas hypomethylation (the Bad Karma epiallele) predicts homeotic transformation, parthenocarpy and marked loss of yield. Loss of Karma methylation and of small RNA in tissue culture contributes to the origin of mantled, while restoration in spontaneous revertants accounts for non-Mendelian inheritance. The ability to predict and cull mantling at the plantlet stage will facilitate the introduction of higher performing clones and optimize environmentally sensitive land resources.
Figures

Comment in
-
Epigenetics: The karma of oil palms.Nature. 2015 Sep 24;525(7570):466-7. doi: 10.1038/nature15216. Epub 2015 Sep 9. Nature. 2015. PMID: 26352472 No abstract available.
-
Epigenetics: Bad Karma reduces palm oil yields.Nat Rev Genet. 2015 Nov;16(11):624-5. doi: 10.1038/nrg4022. Epub 2015 Sep 29. Nat Rev Genet. 2015. PMID: 26416310 No abstract available.
References
-
- Corley RHV. Oil palm. In: Monselise SP, editor. CRC Handbook of fruit set and development. CRC Press; Boca Raton, Florida: 1986. pp. 253–259.
-
- Zeven AC. The ‘mantled’ oil palm (Elaeis guineensis Jacq.) J W Afr Inst Oil Palm Res. 1973;5:31–33.
-
- Mgbeze GC, Iserhienrhien A. Somaclonal variation associated with oil palm (Elaeis guineensis Jacq.) clonal propagation: A review. African Journal of Biotechnology. 2014;13:989–997.
-
- Jaligot E, Rival A, Beule T, Dussert S, Verdeil JL. Somaclonal variation in oil palm (Elaeis guineensis Jacq.): the DNA methylation hypothesis. Pl Cell Rep. 2000;19:684–690. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- SRA/SAMN03569063
- SRA/SAMN03569064
- SRA/SAMN03569065
- SRA/SAMN03569066
- SRA/SAMN03569067
- SRA/SAMN03569068
- SRA/SAMN03569069
- SRA/SAMN03569070
- SRA/SAMN03569071
- SRA/SAMN03569072
- SRA/SAMN03569073
- SRA/SAMN03569074
- SRA/SAMN03569075
- SRA/SAMN03569076
- SRA/SAMN03569077
- SRA/SAMN03569290
- SRA/SAMN03569291
- SRA/SAMN03569292
- SRA/SAMN03569293
- SRA/SAMN03569294
- SRA/SAMN03569295
- SRA/SAMN03569296
- SRA/SAMN03569297
- SRA/SAMN03569298
- SRA/SAMN03569299
- SRA/SAMN03569300
- SRA/SAMN03569301
- SRA/SAMN03569302
- SRA/SAMN03569303
- SRA/SAMN03569304
- SRA/SAMN03569305
- SRA/SAMN03569306
- SRA/SAMN03569307
- SRA/SAMN03569308
- SRA/SAMN03569309
- SRA/SAMN03569310
- SRA/SAMN03569311
- SRA/SAMN03569312
- SRA/SAMN03569313
- SRA/SAMN03569314
- SRA/SAMN03569315
- SRA/SAMN03569316
- SRA/SAMN03569317
- SRA/SAMN03569318
- SRA/SAMN03569319
- SRA/SAMN03569320
- SRA/SAMN03569321
- SRA/SAMN03569322
- SRA/SAMN03569323
- SRA/SAMN03569324
- SRA/SAMN03569325
- SRA/SAMN03569326
- SRA/SAMN03569327
- SRA/SAMN03569328
- SRA/SAMN03569329
- SRA/SAMN03569330
- SRA/SAMN03569331
- SRA/SAMN03569332
- SRA/SAMN03569333
- SRA/SAMN03569334
- SRA/SAMN03569335
- SRA/SAMN03569336
- SRA/SAMN03569337
- SRA/SAMN03569338
- SRA/SAMN03569339
- SRA/SAMN03569340
- SRA/SAMN03569341
- SRA/SAMN03569342
- SRA/SAMN03569343
- SRA/SAMN03569344
- SRA/SAMN03569345
- SRA/SAMN03569346
- SRA/SAMN03569347
- SRA/SAMN03569348
- SRA/SAMN03569349
- SRA/SAMN03569350
- SRA/SAMN03569351
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
