

CAMS-RS: Clustering Algorithm for Large-Scale Mass Spectrometry Data Using Restricted Search Space and Intelligent Random Sampling
- PMID: 26355513
- PMCID: PMC6143137
- DOI: 10.1109/TCBB.2013.152
CAMS-RS: Clustering Algorithm for Large-Scale Mass Spectrometry Data Using Restricted Search Space and Intelligent Random Sampling
Abstract
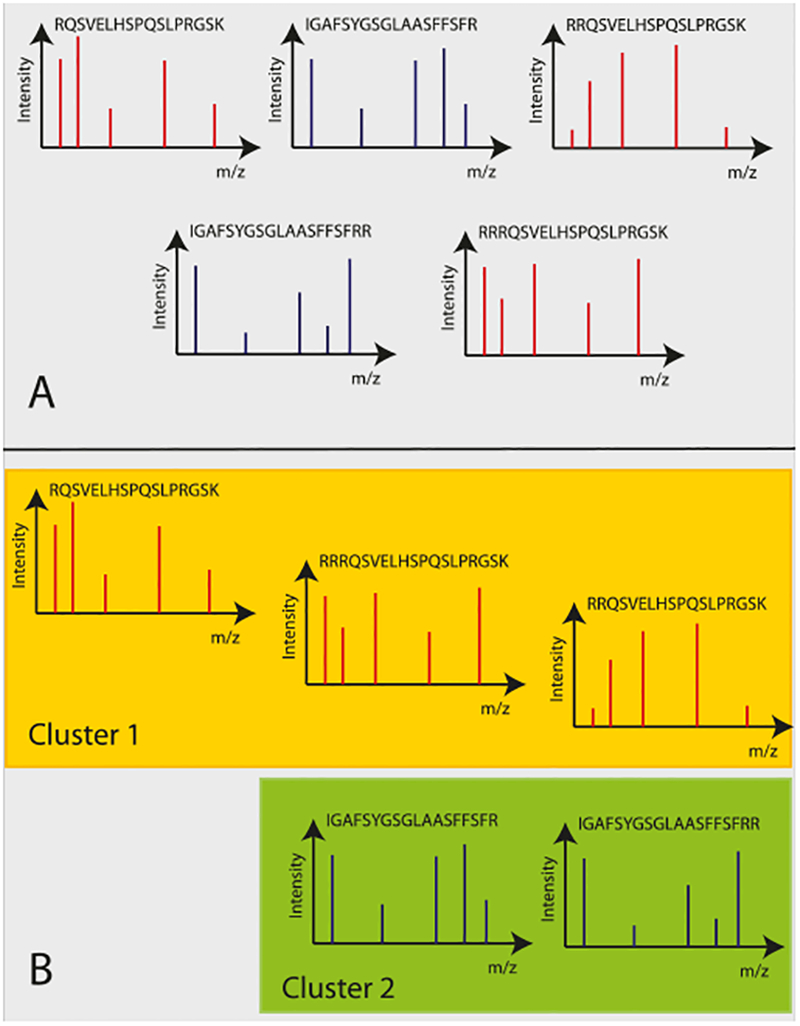
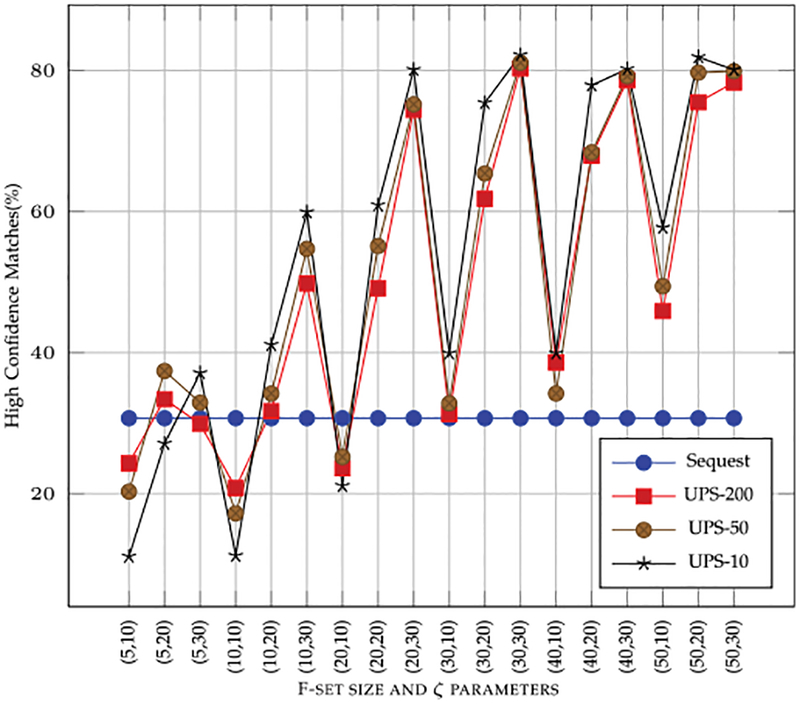
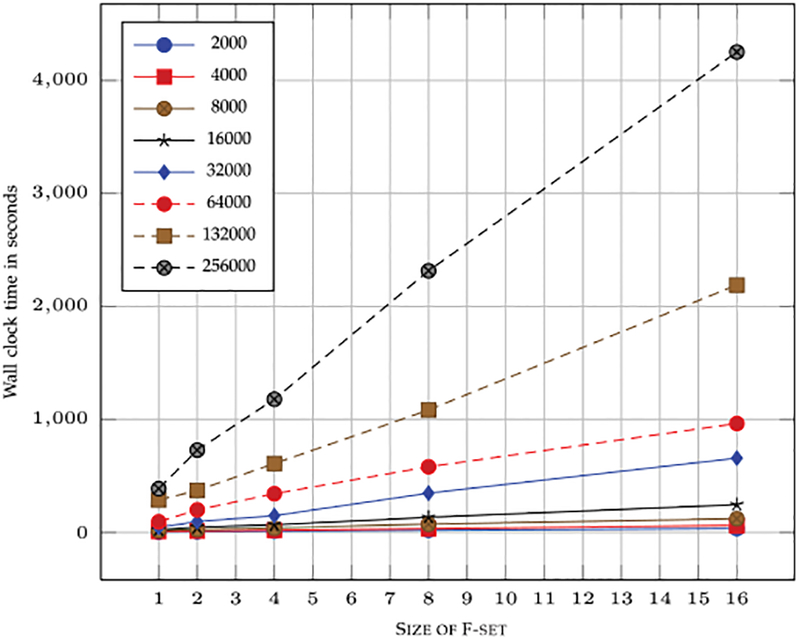
High-throughput mass spectrometers can produce massive amounts of redundant data at an astonishing rate with many of them having poor signal-to-noise (S/N) ratio. These low S/N ratio spectra may not get interpreted using conventional spectra-to-database matching techniques. In this paper, we present an efficient algorithm, CAMS-RS (Clustering Algorithm for Mass Spectra using Restricted Space and Sampling) for clustering of raw mass spectrometry data. CAMS-RS utilizes a novel metric (called F-set) that exploits the temporal and spatial patterns to accurately assess similarity between two given spectra. The F-set similarity metric is independent of the retention time and allows clustering of mass spectrometry data from independent LC-MS/MS runs. A novel restricted search space strategy is devised to limit the comparisons of the number of spectra. An intelligent sampling method is executed on individual bins that allow merging of the results to make the final clusters. Our experiments, using experimentally generated data sets, show that the proposed algorithm is able to cluster spectra with high accuracy and is helpful in interpreting low S/N ratio spectra. The CAMS-RS algorithm is highly scalable with increasing number of spectra and our implementation allows clustering of up to a million spectra within minutes.
Figures











References
-
- Beer I, Barnea E, Ziv T, and Admon A, “Improving Large-Scale Proteomics by Clustering of Mass Spectrometry Data,” Proteomics, vol. 4, no. 4, pp. 950–960, 2004. - PubMed
-
- Bensmail H, Golek J, Moody M, Semmes J, and Haoudi A, “A Novel Approach for Clustering Proteomics Data Using Bayesian Fast Fourier Transform,” Bioinformatics, vol. 21, no. 10, pp. 2210–2224, 2005. - PubMed
-
- De Souza D, Saunders E, McConville M, and Likić V, “Progressive Peak Clustering in Gc-Ms Metabolomic Experiments Applied to Leishmania Parasites,” Bioinformatics, vol. 22, no. 11, pp. 1391–1396, 2006. - PubMed
-
- Dutta D and Chen T, “Speeding up Tandem Mass Spectrometry Database Search: Metric Embeddings and Fast Near Neighbor Search,” Bioinformatics, vol. 23, no. 5, pp. 612–618, 2007. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
