

The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential
- PMID: 26362469
- PMCID: PMC4630564
- DOI: 10.1124/pr.114.009654
The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential
Abstract
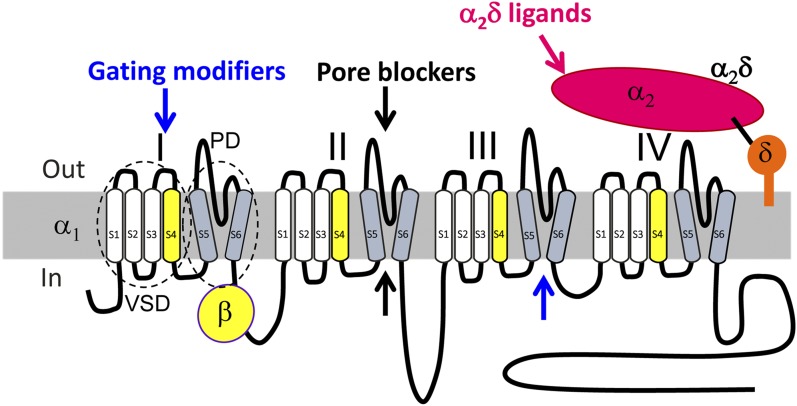
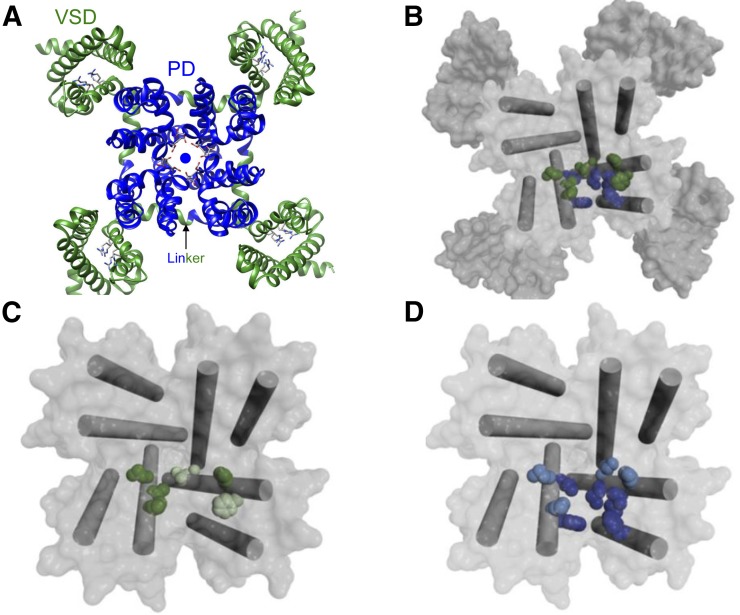
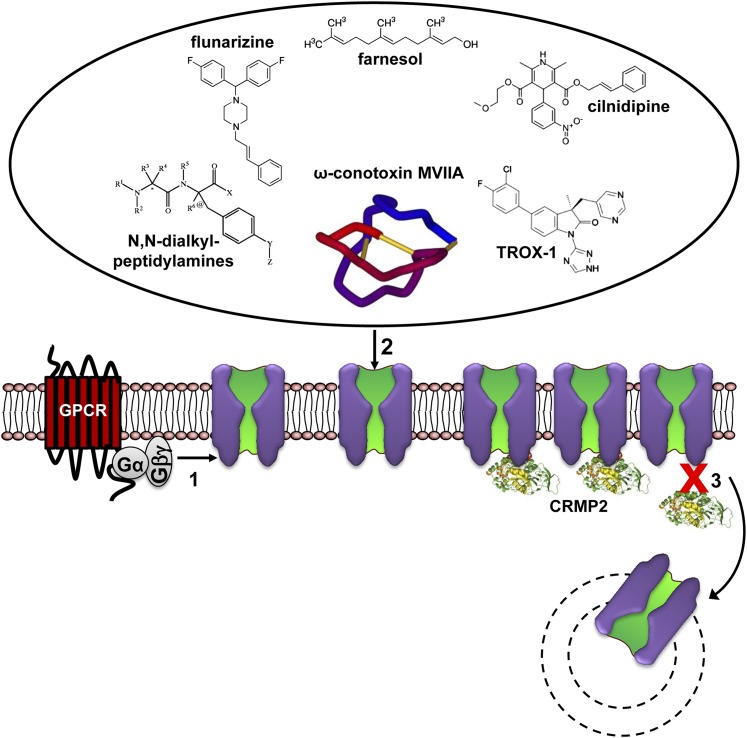
Voltage-gated calcium channels are required for many key functions in the body. In this review, the different subtypes of voltage-gated calcium channels are described and their physiologic roles and pharmacology are outlined. We describe the current uses of drugs interacting with the different calcium channel subtypes and subunits, as well as specific areas in which there is strong potential for future drug development. Current therapeutic agents include drugs targeting L-type Ca(V)1.2 calcium channels, particularly 1,4-dihydropyridines, which are widely used in the treatment of hypertension. T-type (Ca(V)3) channels are a target of ethosuximide, widely used in absence epilepsy. The auxiliary subunit α2δ-1 is the therapeutic target of the gabapentinoid drugs, which are of value in certain epilepsies and chronic neuropathic pain. The limited use of intrathecal ziconotide, a peptide blocker of N-type (Ca(V)2.2) calcium channels, as a treatment of intractable pain, gives an indication that these channels represent excellent drug targets for various pain conditions. We describe how selectivity for different subtypes of calcium channels (e.g., Ca(V)1.2 and Ca(V)1.3 L-type channels) may be achieved in the future by exploiting differences between channel isoforms in terms of sequence and biophysical properties, variation in splicing in different target tissues, and differences in the properties of the target tissues themselves in terms of membrane potential or firing frequency. Thus, use-dependent blockers of the different isoforms could selectively block calcium channels in particular pathologies, such as nociceptive neurons in pain states or in epileptic brain circuits. Of important future potential are selective Ca(V)1.3 blockers for neuropsychiatric diseases, neuroprotection in Parkinson's disease, and resistant hypertension. In addition, selective or nonselective T-type channel blockers are considered potential therapeutic targets in epilepsy, pain, obesity, sleep, and anxiety. Use-dependent N-type calcium channel blockers are likely to be of therapeutic use in chronic pain conditions. Thus, more selective calcium channel blockers hold promise for therapeutic intervention.
Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics.
Figures



References
-
- Abbadie C, McManus OB, Sun SY, Bugianesi RM, Dai G, Haedo RJ, Herrington JB, Kaczorowski GJ, Smith MM, Swensen AM, et al. (2010) Analgesic effects of a substituted N-triazole oxindole (TROX-1), a state-dependent, voltage-gated calcium channel 2 blocker. J Pharmacol Exp Ther 334:545–555. - PubMed
-
- Adams DJ, Smith AB, Schroeder CI, Yasuda T, Lewis RJ. (2003) Omega-conotoxin CVID inhibits a pharmacologically distinct voltage-sensitive calcium channel associated with transmitter release from preganglionic nerve terminals. J Biol Chem 278:4057–4062. - PubMed
-
- Adams ME, Myers RA, Imperial JS, Olivera BM. (1993) Toxityping rat brain calcium channels with omega-toxins from spider and cone snail venoms. Biochemistry 32:12566–12570. - PubMed
-
- Adams PJ, Garcia E, David LS, Mulatz KJ, Spacey SD, Snutch TP. (2009) Ca(V)2.1 P/Q-type calcium channel alternative splicing affects the functional impact of familial hemiplegic migraine mutations: implications for calcium channelopathies. Channels (Austin) 3:110–121. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
