

Hydroxyurea therapy for sickle cell anemia
- PMID: 26366626
- PMCID: PMC5868345
- DOI: 10.1517/14740338.2015.1088827
Hydroxyurea therapy for sickle cell anemia
Abstract
Introduction: Sickle cell anemia (SCA) is a severe, inherited hemoglobin disorder affecting 100,000 persons in the US and millions worldwide. Hydroxyurea, a once daily oral medication, has emerged as the primary disease-modifying therapy for SCA. The accumulated body of evidence over 30 years demonstrates that hydroxyurea is a safe and effective therapy for SCA, but hydroxyurea remains underutilized for a variety of reasons.
Areas covered: In this review, we summarize the available evidence regarding the pharmacology, clinical, and laboratory benefits, and safety of hydroxyurea therapy for the treatment of SCA. The purpose of this review is to provide the reader a comprehensive understanding of hydroxyurea and to reinforce the fact that hydroxyurea is a safe and effective medication for the treatment of SCA.
Expert opinion: In our opinion, hydroxyurea therapy should be considered standard-of-care for SCA, representing an essential component of patient management. Early initiation and broader use of hydroxyurea will alter the natural history of SCA, so affected children can live longer and healthier lives. In addition, hydroxyurea use should be extended to low-resource settings such as sub-Saharan Africa, where the burden of SCA and the need for hydroxyurea is arguably the greatest.
Keywords: fetal hemoglobin; hemoglobinopathies; hydroxyurea; sickle cell anemia.
Conflict of interest statement
Figures
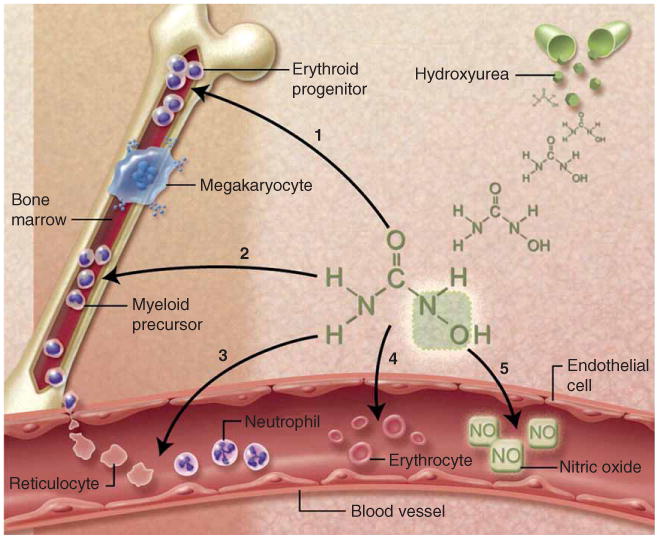
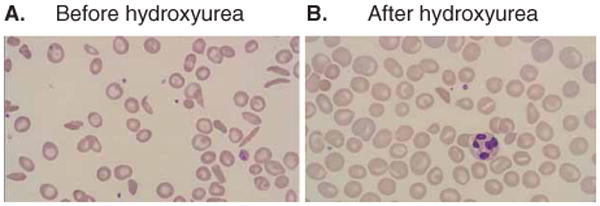
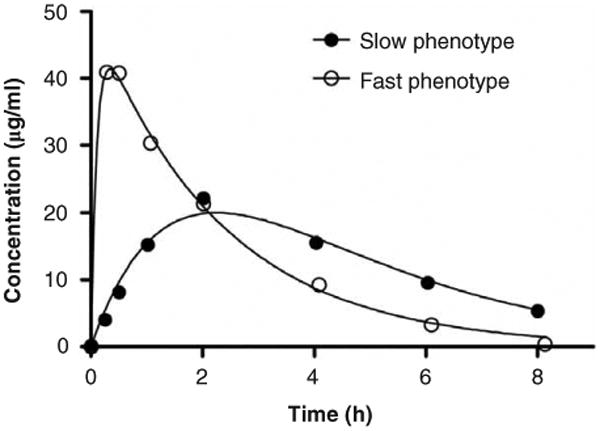
References
-
- Hassell KL. Population Estimates of Sickle Cell Disease in the U.S. Am J Prev Med. 2010;38(4):S512–21. - PubMed
-
- Telfer P, Coen P, Chakravorty S, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica. 2007;92(7):905–12. - PubMed
-
- McGann PT. Sickle cell anemia: an underappreciated and unaddressed contributor to global childhood mortality. J Pediatr. 2014;165(1):18–22. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical