

doi: 10.1038/nmeth.3582.
Epub 2015 Sep 14.
WASP: allele-specific software for robust molecular quantitative trait locus discovery
Affiliations
- PMID: 26366987
- PMCID: PMC4626402
- DOI: 10.1038/nmeth.3582
Item in Clipboard
WASP: allele-specific software for robust molecular quantitative trait locus discovery
Nat Methods.
2015 Nov.
Abstract
Allele-specific sequencing reads provide a powerful signal for identifying molecular quantitative trait loci (QTLs), but they are challenging to analyze and are prone to technical artifacts. Here we describe WASP, a suite of tools for unbiased allele-specific read mapping and discovery of molecular QTLs. Using simulated reads, RNA-seq reads and chromatin immunoprecipitation sequencing (ChIP-seq) reads, we demonstrate that WASP has a low error rate and is far more powerful than existing QTL-mapping approaches.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
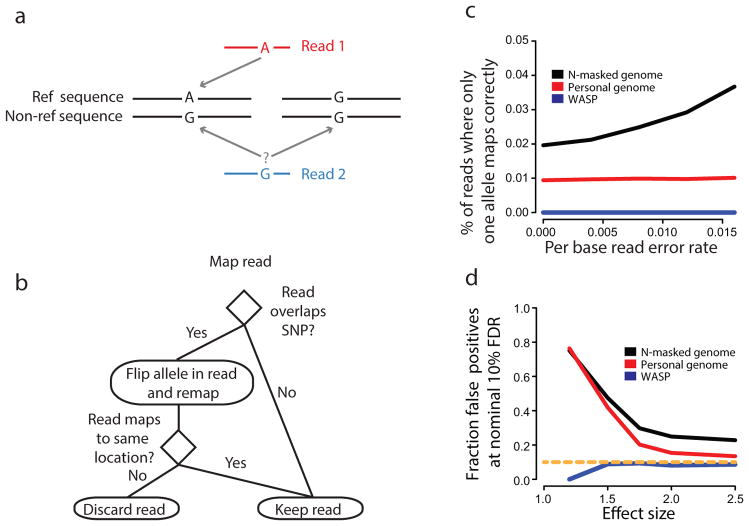
Mapping of allele specific reads. (a) Mapping to ‘personalized’ genomes can result in allelic bias because reads from one allele may not map uniquely. (b) Schematic of mapping pipeline to remove allelic bias. (c) The percentage of simulated 100 bp reads at heterozygous sites where a read with one allele maps correctly and the corresponding read with the other allele does not. Reads were simulated with sequencing errors introduced at several different rates. (d) The fraction of false-positives as a function of the effect size using a nominal Benjamini-Hochberg false-discovery rate of 10%. We simulated 100 bp allele-specific reads under null (odds ratio = 1) and alternative models (odds-ratio > 1) of allelic imbalance at heterozygous sites in the genome. 90% and 10% of sites were assumed to be null and alternative sites respectively. We mapped reads using WASP, personal-genome (AlleleSeq10) or N-masked-genome mapping strategies and called allele-specific sites using a binomial test.
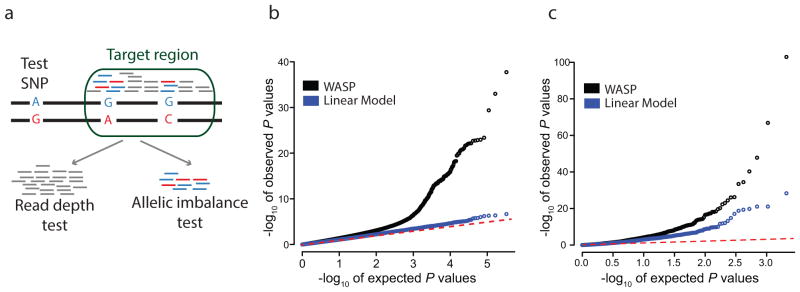
The combined haplotype test and its performance. (a) Schematic of the combined haplotype test. A ‘test SNP’ is tested for association with mapped reads within a ‘target region’. All reads are used by the read depth component of the test; allele-specific reads are used by the allelic imbalance component of the test. (b) Identification of novel QTLs using H3K27ac ChIP-seq data from 10 Yoruba lymphoblastoid cell lines. (c) Identifying European eQTLs from the GEUVADIS consortium using an independent dataset of RNA-seq from 69 Yoruba lymphoblastoid cell lines.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
