

The UK10K project identifies rare variants in health and disease
- PMID: 26367797
- PMCID: PMC4773891
- DOI: 10.1038/nature14962
The UK10K project identifies rare variants in health and disease
Abstract
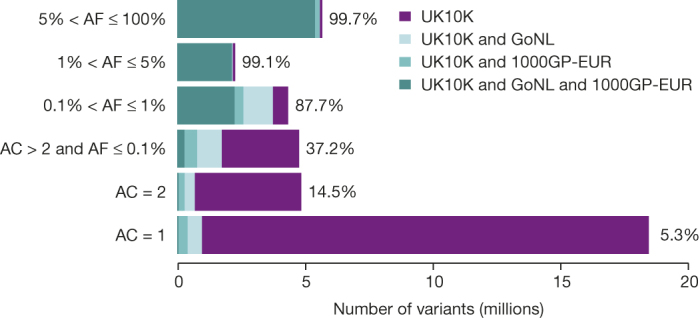
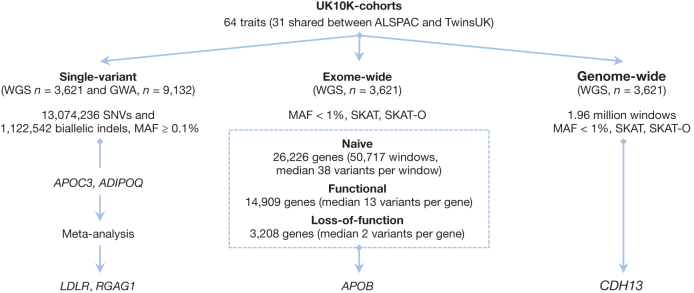
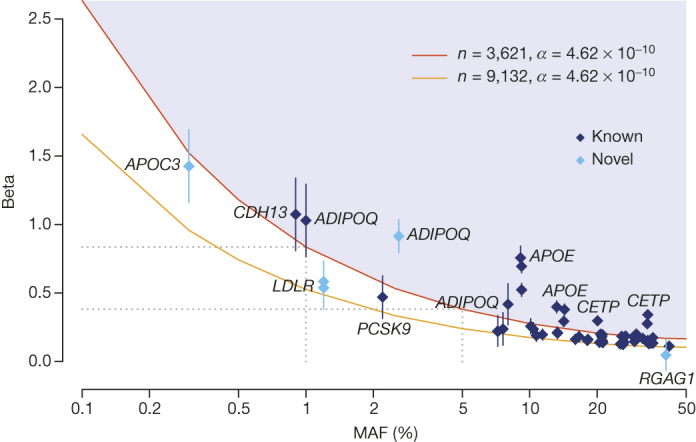
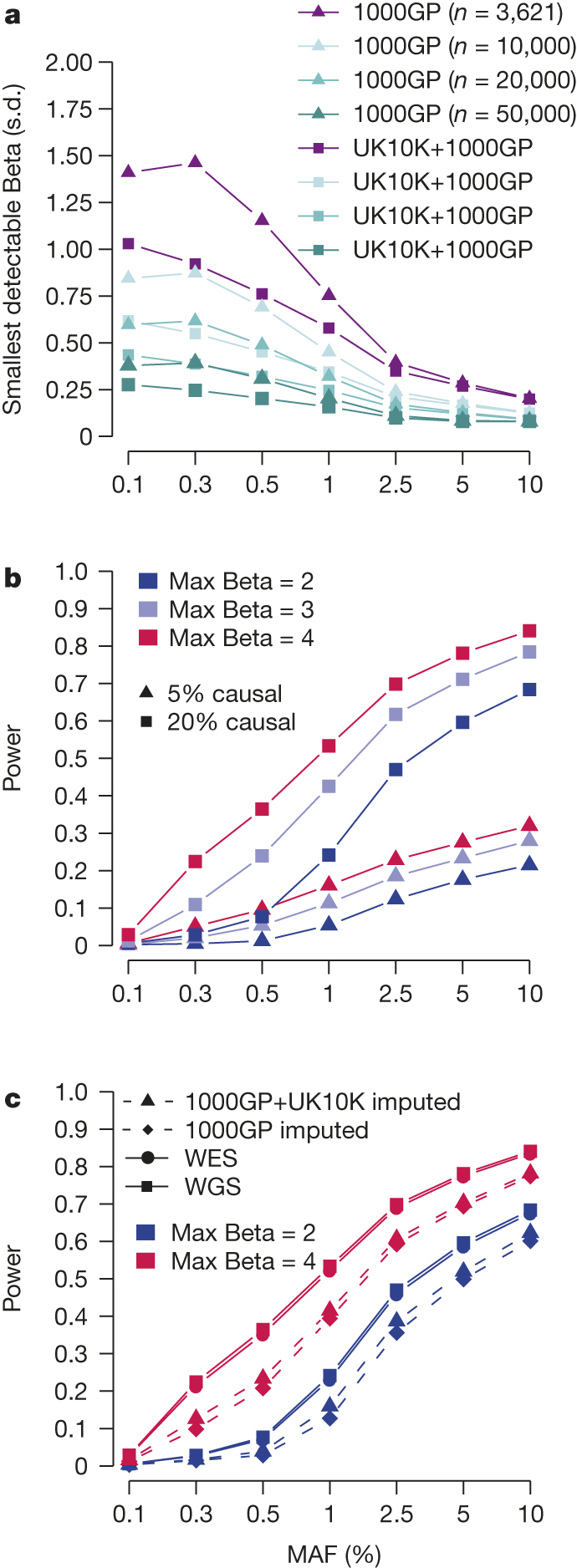
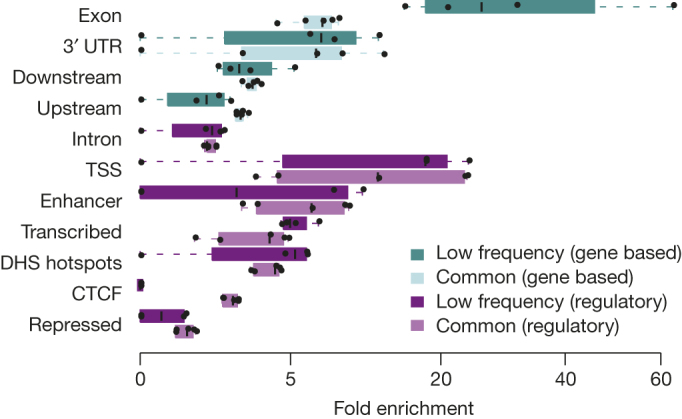
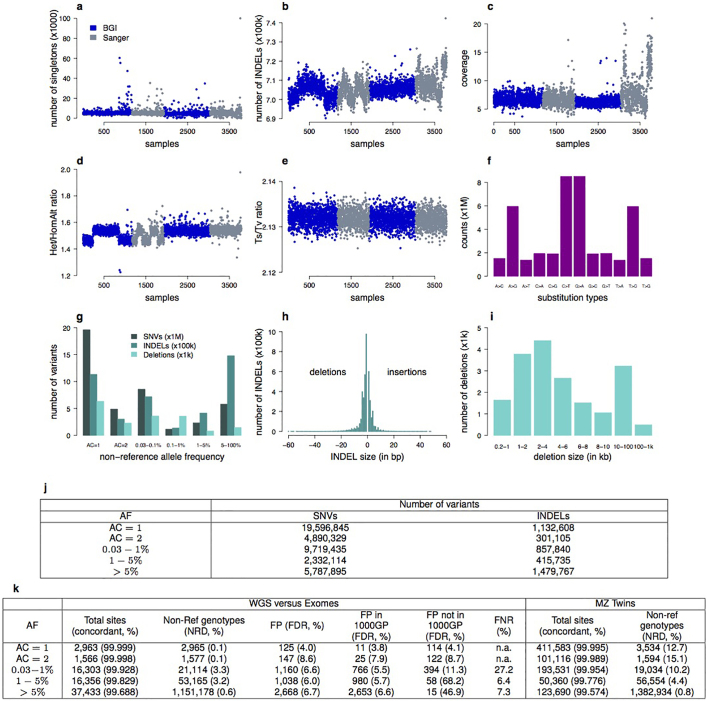
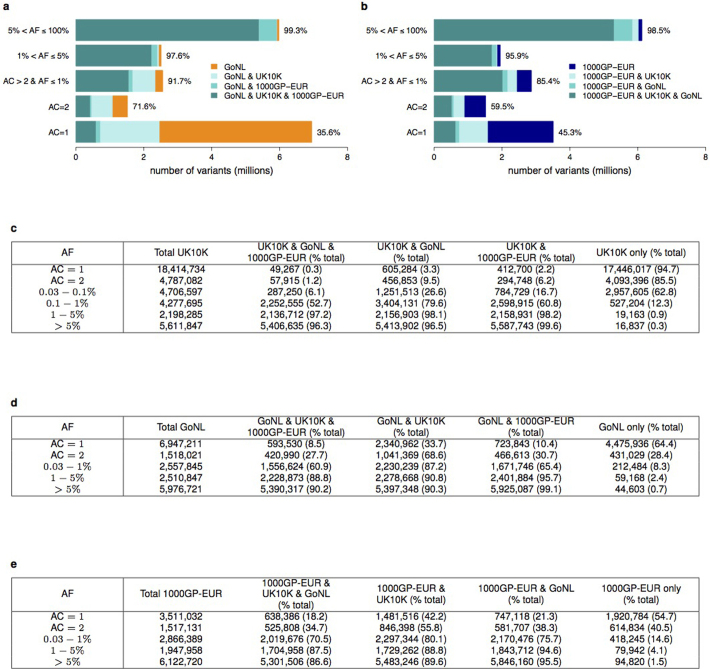
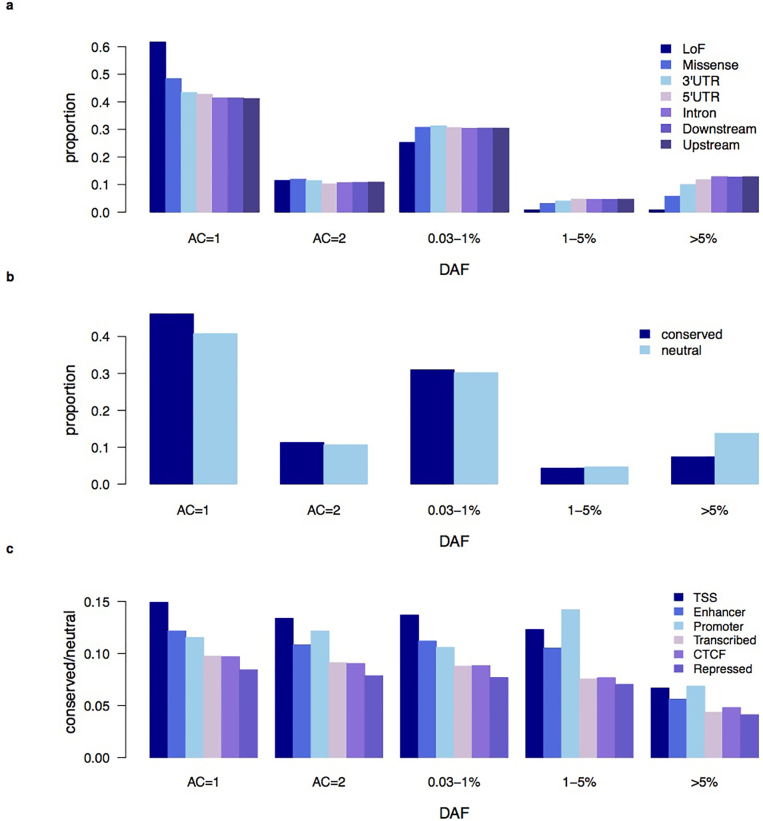
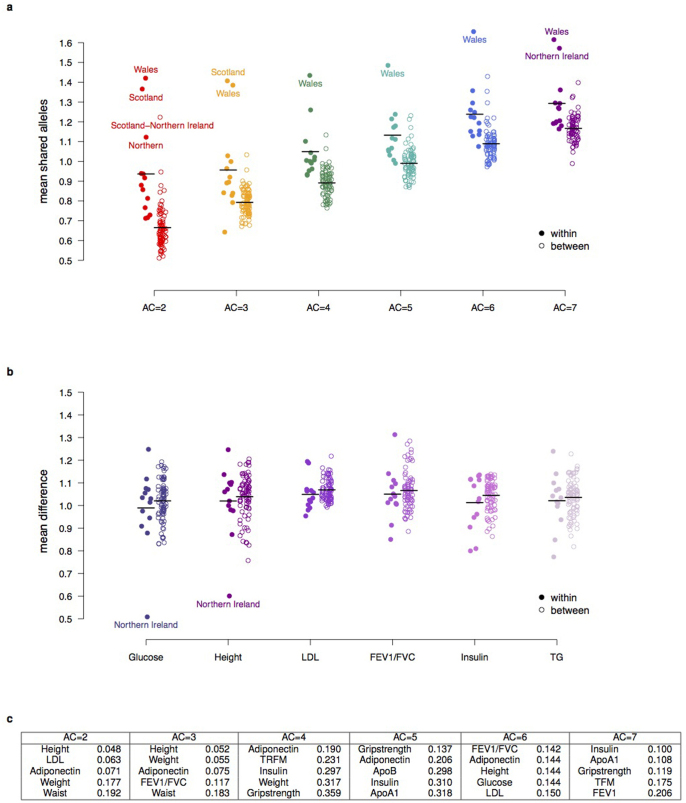
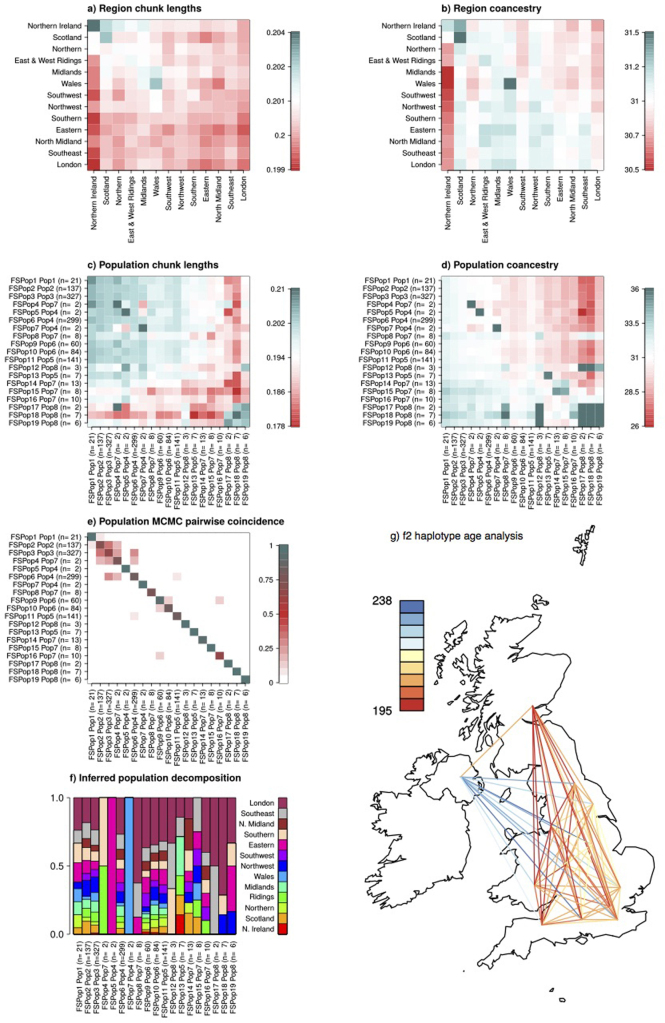
The contribution of rare and low-frequency variants to human traits is largely unexplored. Here we describe insights from sequencing whole genomes (low read depth, 7×) or exomes (high read depth, 80×) of nearly 10,000 individuals from population-based and disease collections. In extensively phenotyped cohorts we characterize over 24 million novel sequence variants, generate a highly accurate imputation reference panel and identify novel alleles associated with levels of triglycerides (APOB), adiponectin (ADIPOQ) and low-density lipoprotein cholesterol (LDLR and RGAG1) from single-marker and rare variant aggregation tests. We describe population structure and functional annotation of rare and low-frequency variants, use the data to estimate the benefits of sequencing for association studies, and summarize lessons from disease-specific collections. Finally, we make available an extensive resource, including individual-level genetic and phenotypic data and web-based tools to facilitate the exploration of association results.
Conflict of interest statement
P.F. is a member of the Scientific Advisory Board of Omicia, Inc.
Figures
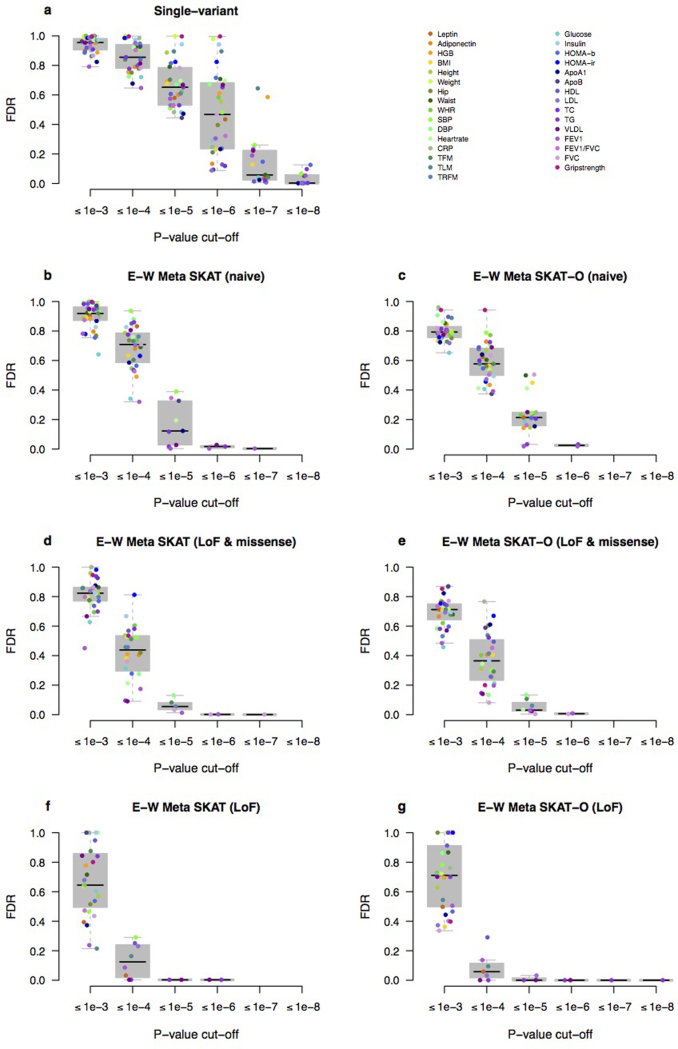
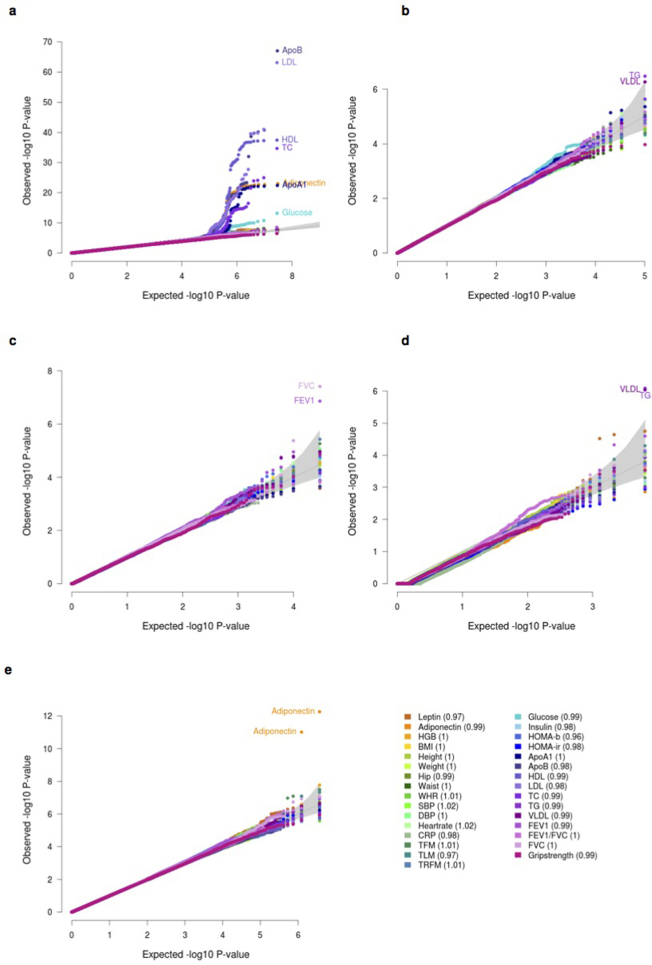
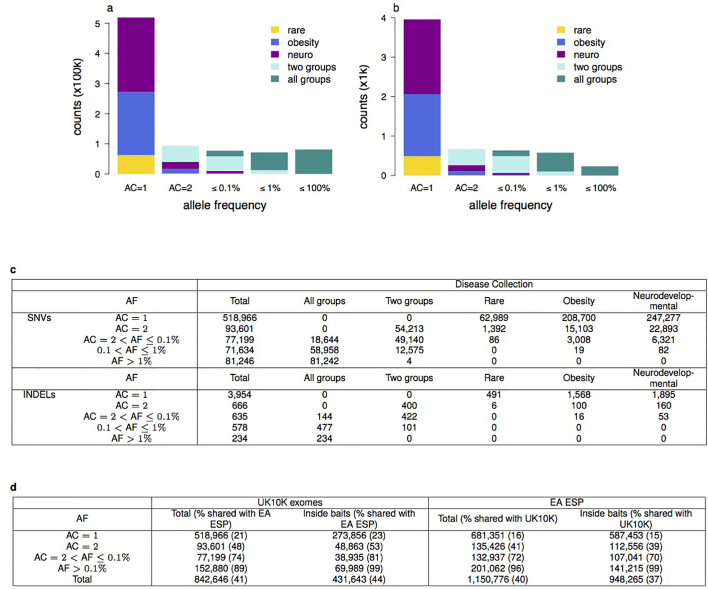
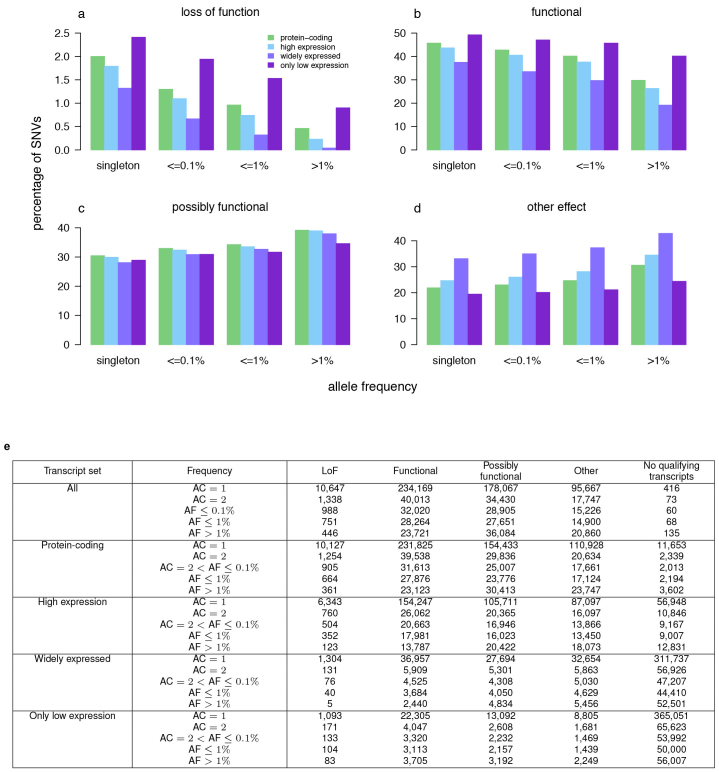
References
Publication types
MeSH terms
Substances
Grants and funding
- G0500870/MRC_/Medical Research Council/United Kingdom
- 104125/WT_/Wellcome Trust/United Kingdom
- 102215/WT_/Wellcome Trust/United Kingdom
- 096599/WT_/Wellcome Trust/United Kingdom
- MC_U106179472/MRC_/Medical Research Council/United Kingdom
- RG/10/17/28553/BHF_/British Heart Foundation/United Kingdom
- MC_UU_12013/1/MRC_/Medical Research Council/United Kingdom
- MR/K013351/1/MRC_/Medical Research Council/United Kingdom
- RG/10/13/28570/BHF_/British Heart Foundation/United Kingdom
- MR/L010305/1/MRC_/Medical Research Council/United Kingdom
- PG/07/045/22690/BHF_/British Heart Foundation/United Kingdom
- RG/08/008/25291/BHF_/British Heart Foundation/United Kingdom
- MC_UU_12013/3/MRC_/Medical Research Council/United Kingdom
- 095564/WT_/Wellcome Trust/United Kingdom
- 104036/WT_/Wellcome Trust/United Kingdom
- MR/J012165/1/MRC_/Medical Research Council/United Kingdom
- SCD/12/CSO_/Chief Scientist Office/United Kingdom
- MC_UU_12012/5/MRC_/Medical Research Council/United Kingdom
- MC_UU_12015/1/MRC_/Medical Research Council/United Kingdom
- WT091310/WT_/Wellcome Trust/United Kingdom
- MC_PC_15018/MRC_/Medical Research Council/United Kingdom
- MC_UU_12015/2/MRC_/Medical Research Council/United Kingdom
- 098498/WT_/Wellcome Trust/United Kingdom
- MC_UU_12013/4/MRC_/Medical Research Council/United Kingdom
- CZD/16/6/4/CSO_/Chief Scientist Office/United Kingdom
- 095515/WT_/Wellcome Trust/United Kingdom
- G0801843/MRC_/Medical Research Council/United Kingdom
- 100574/WT_/Wellcome Trust/United Kingdom
- G0902313/MRC_/Medical Research Council/United Kingdom
- 100140/WT_/Wellcome Trust/United Kingdom
- G0800509/MRC_/Medical Research Council/United Kingdom
- 091551/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
