

DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors
- PMID: 26373278
- PMCID: PMC4573399
- DOI: 10.1016/j.ccell.2015.07.012
DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors
Abstract
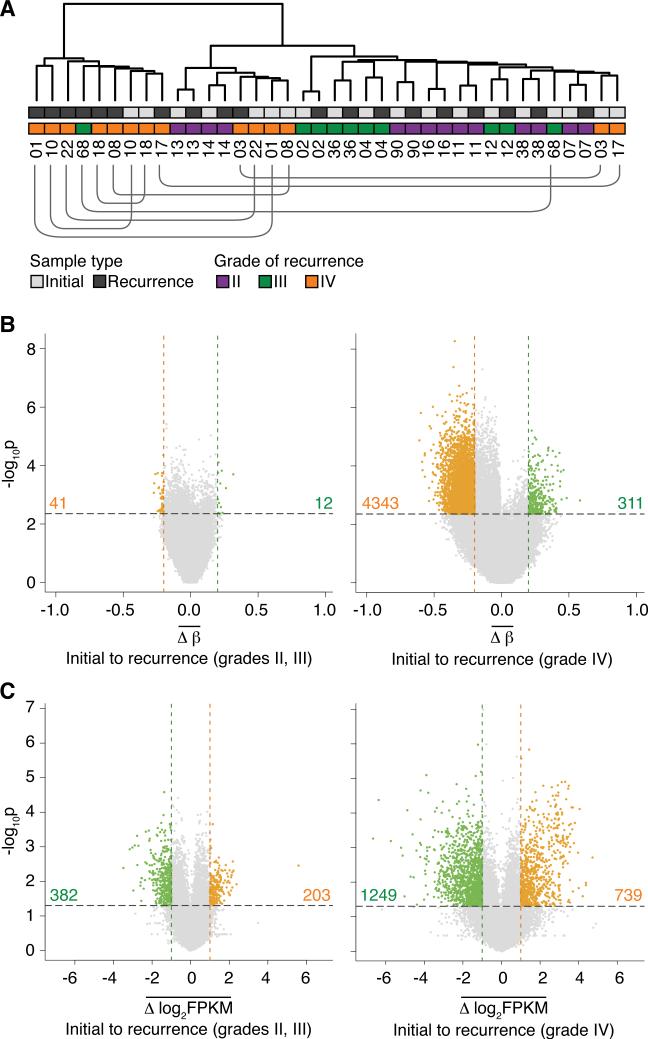
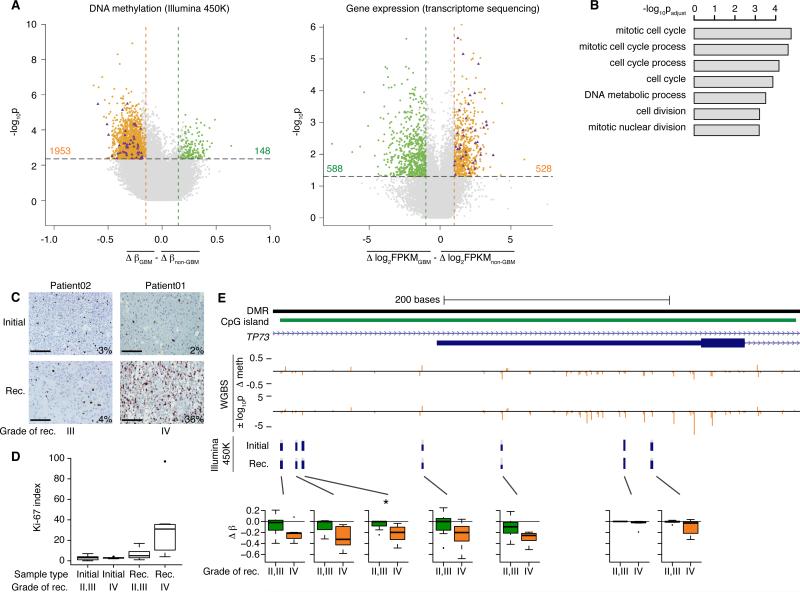
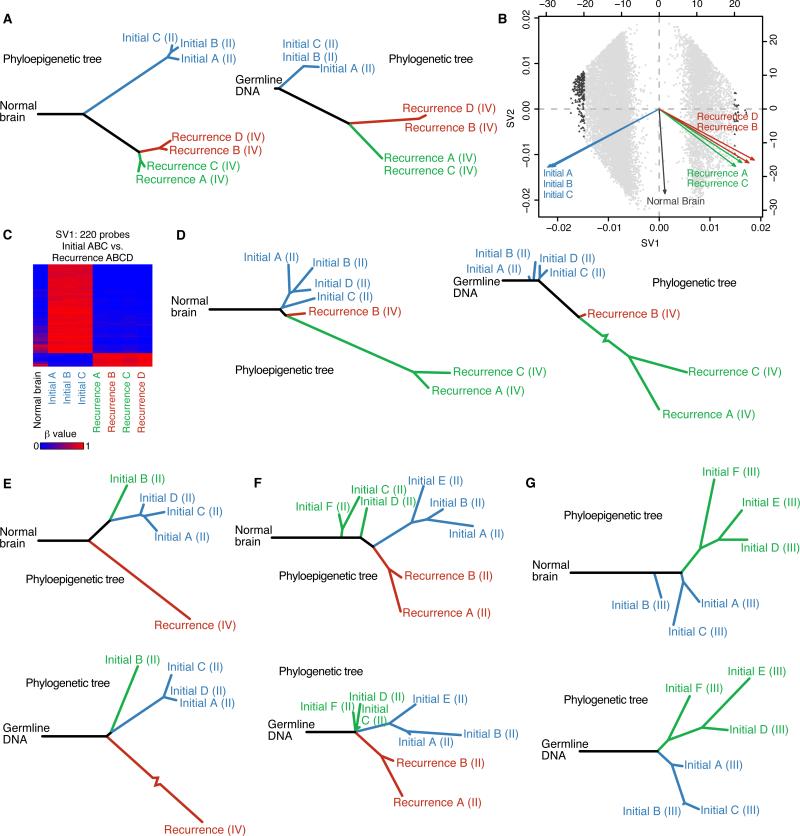
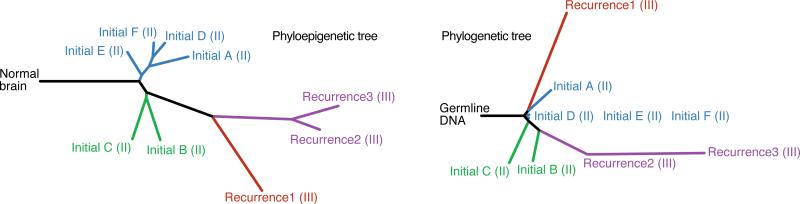
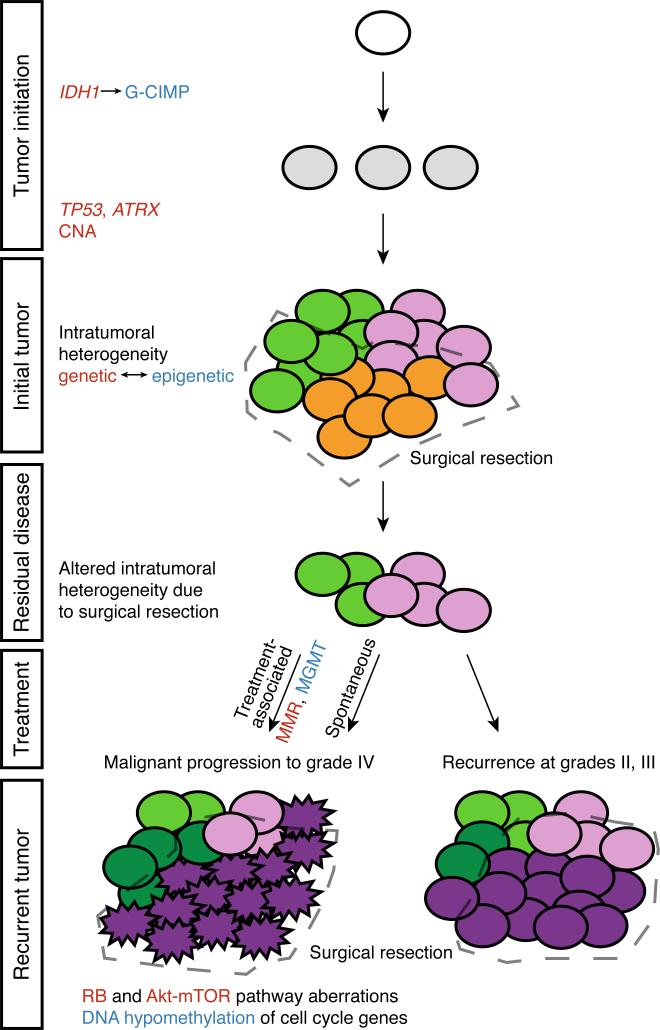
The evolutionary history of tumor cell populations can be reconstructed from patterns of genetic alterations. In contrast to stable genetic events, epigenetic states are reversible and sensitive to the microenvironment, prompting the question whether epigenetic information can similarly be used to discover tumor phylogeny. We examined the spatial and temporal dynamics of DNA methylation in a cohort of low-grade gliomas and their patient-matched recurrences. Genes transcriptionally upregulated through promoter hypomethylation during malignant progression to high-grade glioblastoma were enriched in cell cycle function, evolving in parallel with genetic alterations that deregulate the G1/S cell cycle checkpoint. Moreover, phyloepigenetic relationships robustly recapitulated phylogenetic patterns inferred from somatic mutations. These findings highlight widespread co-dependency of genetic and epigenetic events throughout brain tumor evolution.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
Comment in
-
The Amazing and Deadly Glioma Race.Cancer Cell. 2015 Sep 14;28(3):275-7. doi: 10.1016/j.ccell.2015.08.010. Cancer Cell. 2015. PMID: 26373273
References
-
- Baeza N, Weller M, Yonekawa Y, Kleihues P, Ohgaki H. PTEN methylation and expression in glioblastomas. Acta Neuropathol. 2003;106:479–485. - PubMed
-
- Banine F, Bartlett C, Gunawardena R, Muchardt C, Yaniv M, Knudsen ES, Weissman BE, Sherman LS. SWI/SNF chromatin-remodeling factors induce changes in DNA methylation to promote transcriptional activation. Cancer research. 2005;65:3542–3547. - PubMed
-
- Brocks D, Assenov Y, Minner S, Bogatyrova O, Simon R, Koop C, Oakes C, Zucknick M, Lipka DB, Weischenfeldt J, et al. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell reports. 2014;8:798–806. - PubMed
Publication types
MeSH terms
Grants and funding
- T32 GM067547/GM/NIGMS NIH HHS/United States
- P01 CA118816/CA/NCI NIH HHS/United States
- R01 CA169316/CA/NCI NIH HHS/United States
- T32GM008568/GM/NIGMS NIH HHS/United States
- R25 NS070680/NS/NINDS NIH HHS/United States
- R01 CA163336/CA/NCI NIH HHS/United States
- R25NS070680/NS/NINDS NIH HHS/United States
- P30 CA082103/CA/NCI NIH HHS/United States
- T32GM067547/GM/NIGMS NIH HHS/United States
- R01CA169316/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- P50 CA097257/CA/NCI NIH HHS/United States
- P50CA097257/CA/NCI NIH HHS/United States
- T32 GM008568/GM/NIGMS NIH HHS/United States
- R21 NS093654/NS/NINDS NIH HHS/United States
- R01CA163336/CA/NCI NIH HHS/United States
- P30CA82103/CA/NCI NIH HHS/United States
- 1T32CA15102201/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical