

Overexpressing superoxide dismutase 2 induces a supernormal cardiac function by enhancing redox-dependent mitochondrial function and metabolic dilation
- PMID: 26374996
- PMCID: PMC4641048
- DOI: 10.1016/j.yjmcc.2015.09.001
Overexpressing superoxide dismutase 2 induces a supernormal cardiac function by enhancing redox-dependent mitochondrial function and metabolic dilation
Abstract
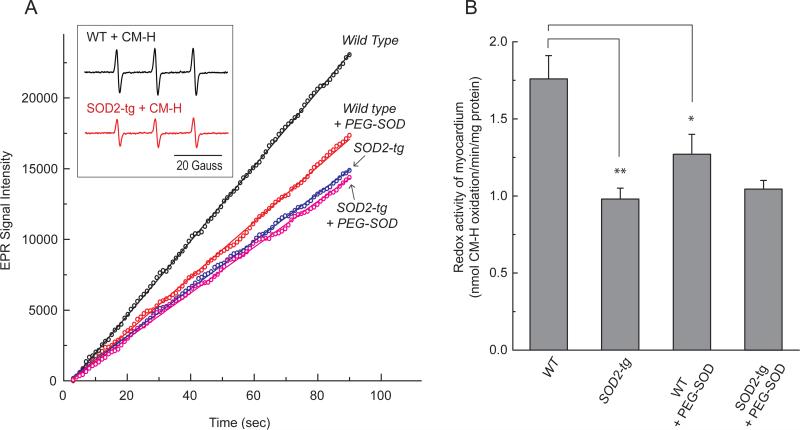
During heightened cardiac work, O2 consumption by the heart benefits energy production via mitochondria. However, some electrons leak from the respiratory chain and yield superoxide, which is rapidly metabolized into H2O2 by SOD2. To understand the systemic effects of the metabolic dilator, H2O2, we studied mice with cardiac-specific SOD2 overexpression (SOD2-tg), which increases the H2O2 produced by cardiac mitochondria. Contrast echocardiography was employed to evaluate cardiac function, indicating that SOD2-tg had a significantly greater ejection fraction and a lower mean arterial pressure (MAP) that was partially normalized by intravenous injection of catalase. Norepinephrine-mediated myocardial blood flow (MBF) was significantly enhanced in SOD2-tg mice. Coupling of MBF to the double product (Heart Rate×MAP) was increased in SOD2-tg mice, indicating that the metabolic dilator, "spilled" over, inducing systemic vasodilation. The hypothesis that SOD2 overexpression effectively enhances mitochondrial function was further evaluated. Mitochondria of SOD2-tg mice had a decreased state 3 oxygen consumption rate, but maintained the same ATP production flux under the basal and L-NAME treatment conditions, indicating a higher bioenergetic efficiency. SOD2-tg mitochondria produced less superoxide, and had lower redox activity in converting cyclic hydroxylamine to stable nitroxide, and a lower GSSG concentration. EPR analysis of the isolated mitochondria showed a significant decrease in semiquinones at the SOD2-tg Qi site. These results support a more reductive physiological setting in the SOD2-tg murine heart. Cardiac mitochondria exhibited no significant differences in the respiratory control index between WT and SOD2-tg. We conclude that SOD2 overexpression in myocytes enhances mitochondrial function and metabolic vasodilation, leading to a phenotype of supernormal cardiac function.
Keywords: Bioenergetics; Cardiac function; Metabolic dilation; Mitochondria; Redox regulation; Superoxide dismutase 2 (SOD2); Transgenic mice.
Copyright © 2015 Elsevier Ltd. All rights reserved.
Figures









References
-
- Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93:573–80. Epub 2003 Aug 14. - PubMed
-
- Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H2023–31. Epub 07 Jan 19. - PubMed
-
- Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, et al. H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ Res. 2012;110:471–80. doi: 10.1161/CIRCRESAHA.111.258871. Epub 2011 Dec 8. - PMC - PubMed
-
- Saitoh S, Zhang C, Tune JD, Potter B, Kiyooka T, Rogers PA, et al. Hydrogen peroxide: a feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler Thromb Vasc Biol. 2006;26:2614–21. Epub 006 Oct 5. - PubMed
-
- Rogers PA, Chilian WM, Bratz IN, Bryan RM, Jr., Dick GM. H2O2 activates redox- and 4-aminopyridine-sensitive Kv channels in coronary vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2007;292:H1404–11. Epub 2006 Oct 27. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
