

Assessing DNA methylation in the developing human intestinal epithelium: potential link to inflammatory bowel disease
- PMID: 26376367
- PMCID: PMC4854977
- DOI: 10.1038/mi.2015.88
Assessing DNA methylation in the developing human intestinal epithelium: potential link to inflammatory bowel disease
Abstract
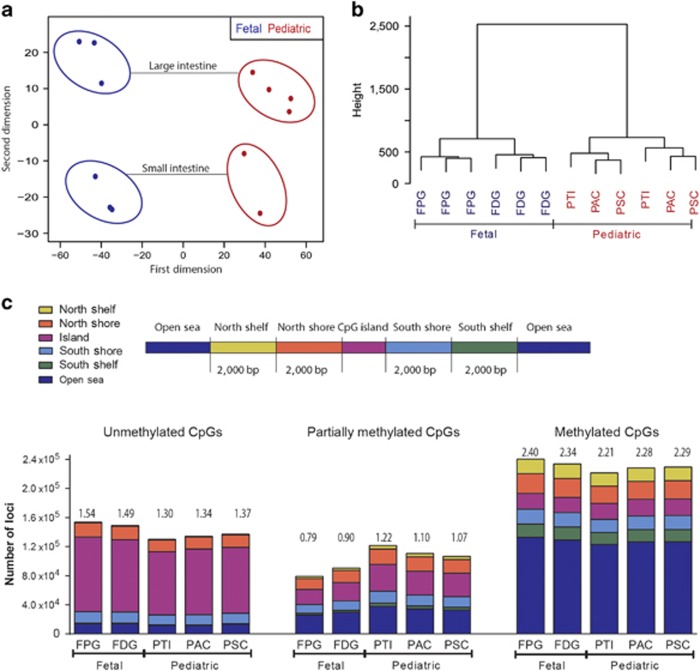
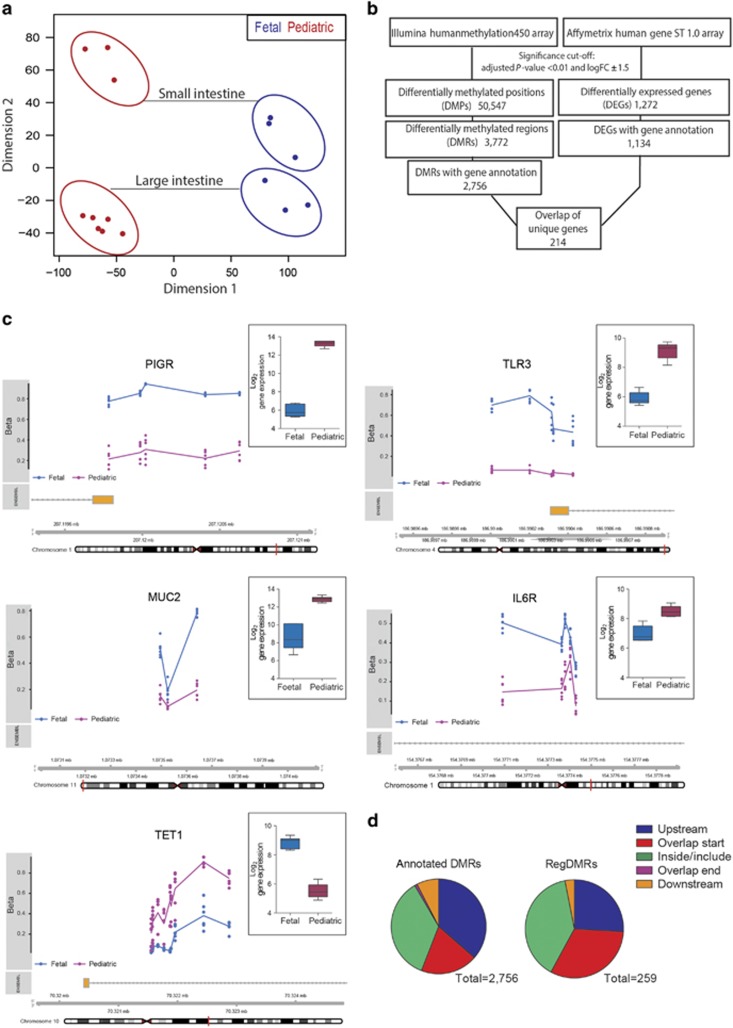
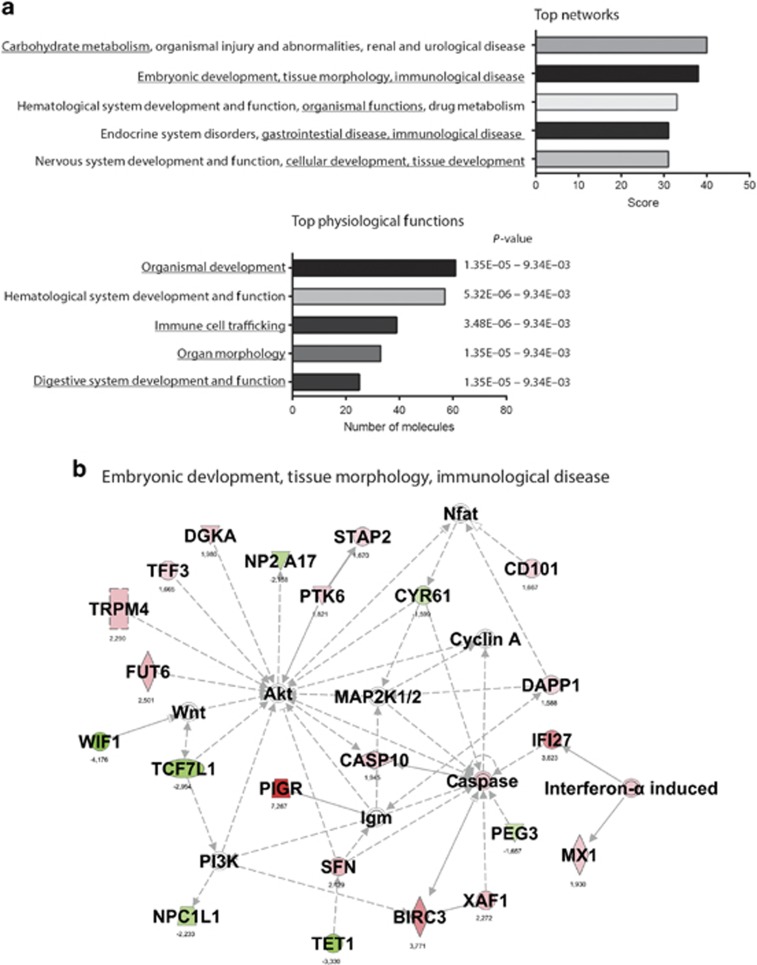
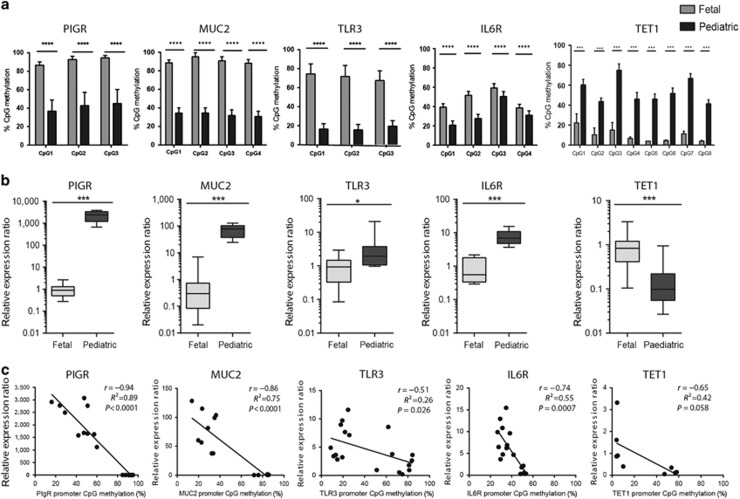
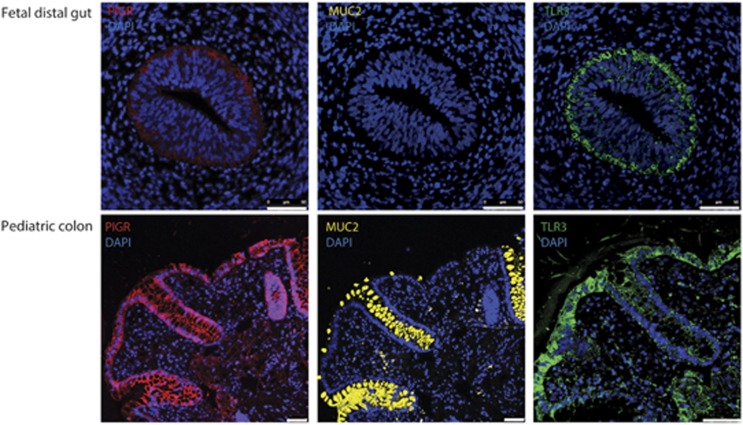
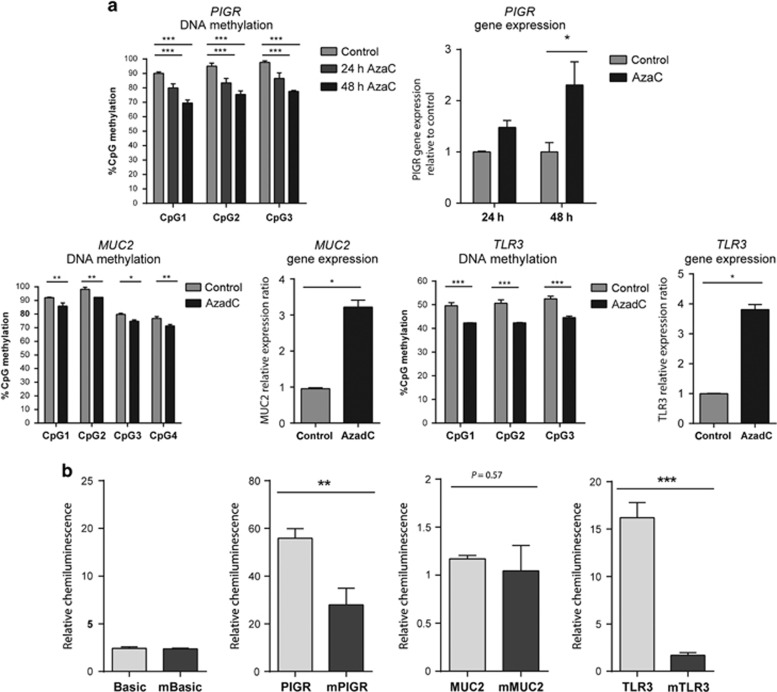
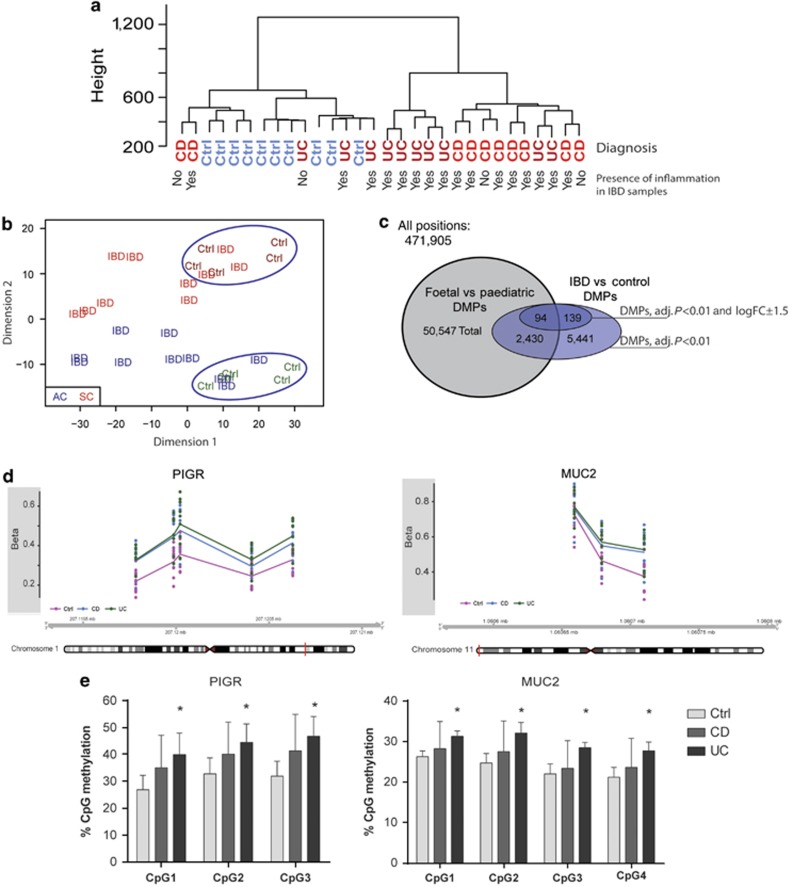
DNA methylation is one of the major epigenetic mechanisms implicated in regulating cellular development and cell-type-specific gene expression. Here we performed simultaneous genome-wide DNA methylation and gene expression analysis on purified intestinal epithelial cells derived from human fetal gut, healthy pediatric biopsies, and children newly diagnosed with inflammatory bowel disease (IBD). Results were validated using pyrosequencing, real-time PCR, and immunostaining. The functional impact of DNA methylation changes on gene expression was assessed by employing in-vitro assays in intestinal cell lines. DNA methylation analyses allowed identification of 214 genes for which expression is regulated via DNA methylation, i.e. regulatory differentially methylated regions (rDMRs). Pathway and functional analysis of rDMRs suggested a critical role for DNA methylation in regulating gene expression and functional development of the human intestinal epithelium. Moreover, analysis performed on intestinal epithelium of children newly diagnosed with IBD revealed alterations in DNA methylation within genomic loci, which were found to overlap significantly with those undergoing methylation changes during intestinal development. Our study provides novel insights into the physiological role of DNA methylation in regulating functional maturation of the human intestinal epithelium. Moreover, we provide data linking developmentally acquired alterations in the DNA methylation profile to changes seen in pediatric IBD.
Figures
References
-
- Peterson, L.W. & Artis, D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 14, 141–153 (2014). - PubMed
-
- Renz, H., Brandtzaeg, P. & Hornef, M. The impact of perinatal immune development on mucosal homeostasis and chronic inflammation. Nat. Rev. Immunol. 12, 9–23 (2012). - PubMed
-
- Dupaul-Chicoine, J., Dagenais, M. & Saleh, M. Crosstalk between the intestinal microbiota and the innate immune system in intestinal homeostasis and inflammatory bowel disease. Inflamm. Bowel Dis. 19, 2227–2237 (2013). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
