

Structural Basis of Neuronal Nitric-oxide Synthase Interaction with Dystrophin Repeats 16 and 17
- PMID: 26378238
- PMCID: PMC4705953
- DOI: 10.1074/jbc.M115.680660
Structural Basis of Neuronal Nitric-oxide Synthase Interaction with Dystrophin Repeats 16 and 17
Abstract
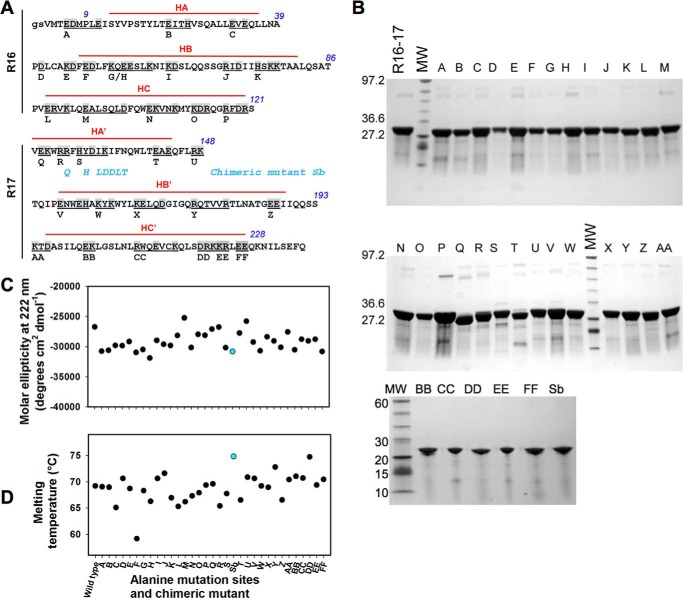
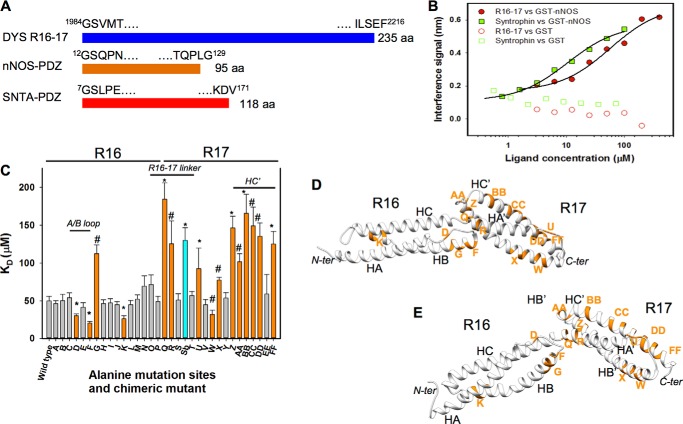
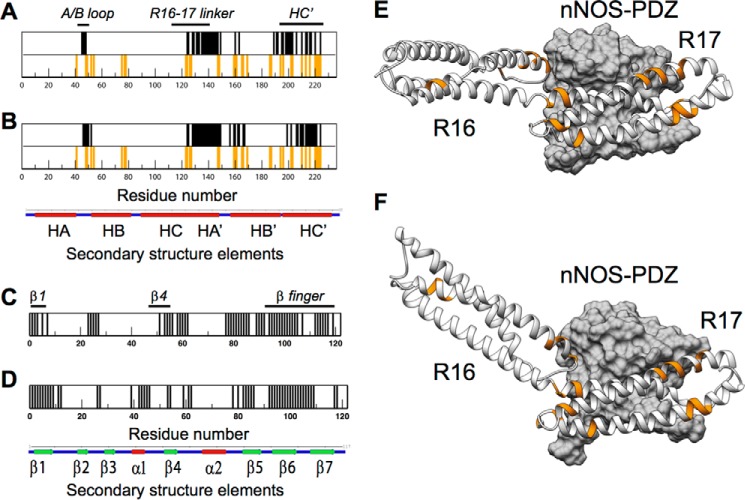
Duchenne muscular dystrophy is a lethal genetic defect that is associated with the absence of dystrophin protein. Lack of dystrophin protein completely abolishes muscular nitric-oxide synthase (NOS) function as a regulator of blood flow during muscle contraction. In normal muscles, nNOS function is ensured by its localization at the sarcolemma through an interaction of its PDZ domain with dystrophin spectrin-like repeats R16 and R17. Early studies suggested that repeat R17 is the primary site of interaction but ignored the involved nNOS residues, and the R17 binding site has not been described at an atomic level. In this study, we characterized the specific amino acids involved in the binding site of nNOS-PDZ with dystrophin R16-17 using combined experimental biochemical and structural in silico approaches. First, 32 alanine-scanning mutagenesis variants of dystrophin R16-17 indicated the regions where mutagenesis modified the affinity of the dystrophin interaction with the nNOS-PDZ. Second, using small angle x-ray scattering-based models of dystrophin R16-17 and molecular docking methods, we generated atomic models of the dystrophin R16-17·nNOS-PDZ complex that correlated well with the alanine scanning identified regions of dystrophin. The structural regions constituting the dystrophin interaction surface involve the A/B loop and the N-terminal end of helix B of repeat R16 and the N-terminal end of helix A' and a small fraction of helix B' and a large part of the helix C' of repeat R17. The interaction surface of nNOS-PDZ involves its main β-sheet and its specific C-terminal β-finger.
Keywords: dystrophin; molecular dynamics; muscular dystrophy; nitric-oxide synthase; site-directed mutagenesis.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures



References
-
- Flanigan K. M., Dunn D. M., von Niederhausern A., Soltanzadeh P., Gappmaier E., Howard M. T., Sampson J. B., Mendell J. R., Wall C., King W. M., Pestronk A., Florence J. M., Connolly A. M., Mathews K. D., Stephan C. M., Laubenthal K. S., Wong B. L., Morehart P. J., Meyer A., Finkel R. S., Bonnemann C. G., Medne L., Day J. W., Dalton J. C., Margolis M. K., Hinton V. J., United Dystrophinopathy Project Consortium, and Weiss R. B. (2009) Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum. Mutat. 30, 1657–1666 - PMC - PubMed
-
- Tuffery-Giraud S., Béroud C., Leturcq F., Yaou R. B., Hamroun D., Michel-Calemard L., Moizard M. P., Bernard R., Cossée M., Boisseau P., Blayau M., Creveaux I., Guiochon-Mantel A., de Martinville B., Philippe C., Monnier N., Bieth E., Khau Van Kien P., Desmet F. O., Humbertclaude V., Kaplan J. C., Chelly J., and Claustres M. (2009) Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum. Mutat. 30, 934–945 - PubMed
-
- Fairclough R. J., Wood M. J., and Davies K. E. (2013) Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat. Rev. Genet. 14, 373–378 - PubMed
-
- Wein N., Alfano L., and Flanigan K. M. (2015) Genetics and emerging treatments for Duchenne and Becker muscular dystrophy. Pediatr. Clin. N. Am. 62, 723–742 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
