

Metabolome of human gut microbiome is predictive of host dysbiosis
- PMID: 26380076
- PMCID: PMC4570295
- DOI: 10.1186/s13742-015-0084-3
Metabolome of human gut microbiome is predictive of host dysbiosis
Abstract
Background: Humans live in constant and vital symbiosis with a closely linked bacterial ecosystem called the microbiome, which influences many aspects of human health. When this microbial ecosystem becomes disrupted, the health of the human host can suffer; a condition called dysbiosis. However, the community compositions of human microbiomes also vary dramatically from individual to individual, and over time, making it difficult to uncover the underlying mechanisms linking the microbiome to human health. We propose that a microbiome's interaction with its human host is not necessarily dependent upon the presence or absence of particular bacterial species, but instead is dependent on its community metabolome; an emergent property of the microbiome.
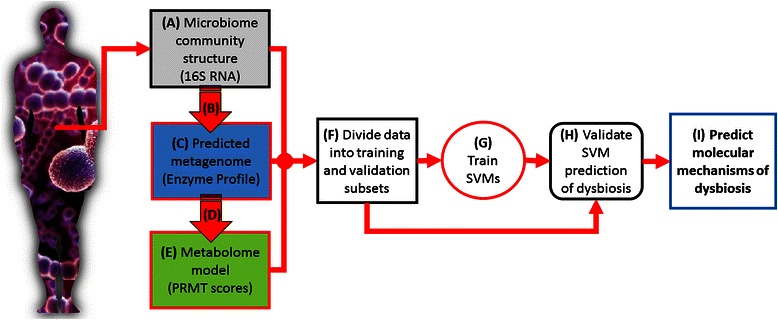
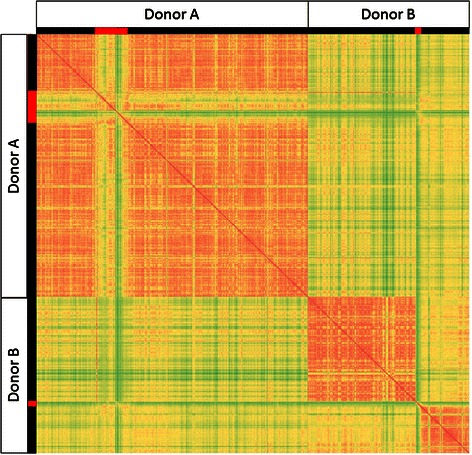
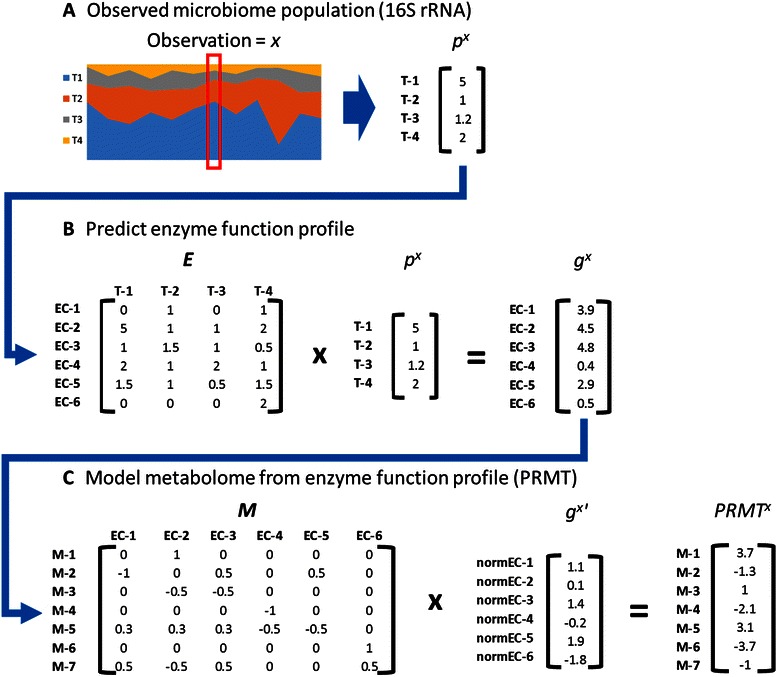
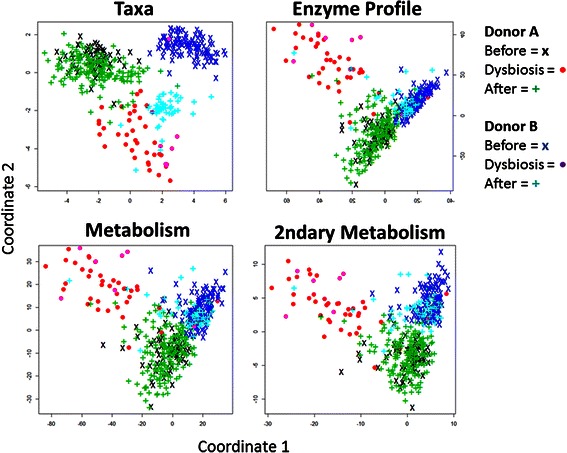
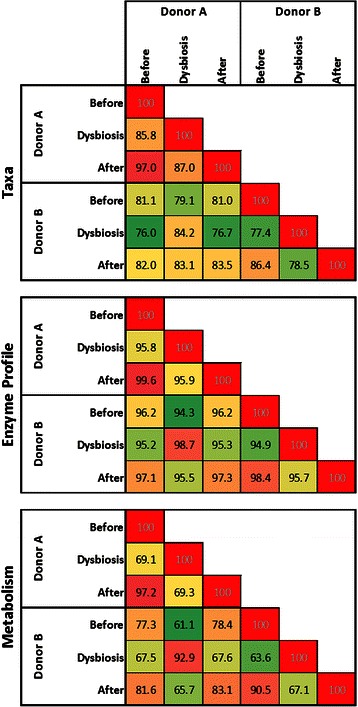
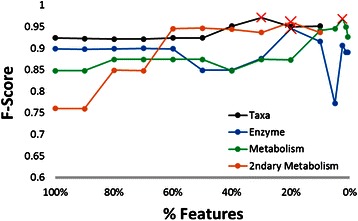
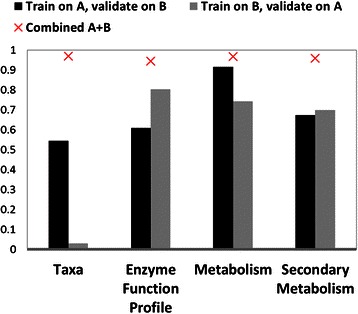
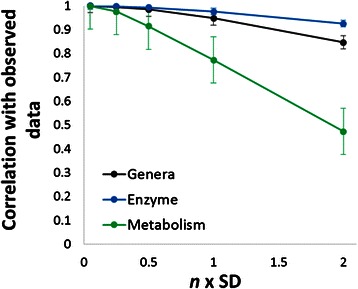
Results: Using data from a previously published, longitudinal study of microbiome populations of the human gut, we extrapolated information about microbiome community enzyme profiles and metabolome models. Using machine learning techniques, we demonstrated that the aggregate predicted community enzyme function profiles and modeled metabolomes of a microbiome are more predictive of dysbiosis than either observed microbiome community composition or predicted enzyme function profiles.
Conclusions: Specific enzyme functions and metabolites predictive of dysbiosis provide insights into the molecular mechanisms of microbiome-host interactions. The ability to use machine learning to predict dysbiosis from microbiome community interaction data provides a potentially powerful tool for understanding the links between the human microbiome and human health, pointing to potential microbiome-based diagnostics and therapeutic interventions.
Keywords: Dysbiosis; Gut microbiome; Human microbiome; Machine learning; Metabolome modeling; Metagenomics; Microbial communities.
Figures
Similar articles
-
A meta-analysis study of the robustness and universality of gut microbiome-metabolome associations.Microbiome. 2021 Oct 12;9(1):203. doi: 10.1186/s40168-021-01149-z. Microbiome. 2021. PMID: 34641974 Free PMC article.
-
Microbiome-metabolome reveals the contribution of gut-kidney axis on kidney disease.J Transl Med. 2019 Jan 3;17(1):5. doi: 10.1186/s12967-018-1756-4. J Transl Med. 2019. PMID: 30602367 Free PMC article. Review.
-
Application of metagenomics in the human gut microbiome.World J Gastroenterol. 2015 Jan 21;21(3):803-14. doi: 10.3748/wjg.v21.i3.803. World J Gastroenterol. 2015. PMID: 25624713 Free PMC article. Review.
-
Mechanistic links between gut microbial community dynamics, microbial functions and metabolic health.World J Gastroenterol. 2014 Nov 28;20(44):16498-517. doi: 10.3748/wjg.v20.i44.16498. World J Gastroenterol. 2014. PMID: 25469018 Free PMC article. Review.
-
A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice.ISME J. 2013 Oct;7(10):1933-43. doi: 10.1038/ismej.2013.89. Epub 2013 Jun 6. ISME J. 2013. PMID: 23739052 Free PMC article.
Cited by
-
Pivotal Dominant Bacteria Ratio and Metabolites Related to Healthy Body Index Revealed by Intestinal Microbiome and Metabolomics.Indian J Microbiol. 2022 Mar;62(1):130-141. doi: 10.1007/s12088-021-00989-5. Epub 2021 Nov 9. Indian J Microbiol. 2022. PMID: 35068612 Free PMC article.
-
Profiling gut microbiota and bile acid metabolism in critically ill children.Sci Rep. 2022 Jun 21;12(1):10432. doi: 10.1038/s41598-022-13640-0. Sci Rep. 2022. PMID: 35729169 Free PMC article.
-
Deep in the Bowel: Highly Interpretable Neural Encoder-Decoder Networks Predict Gut Metabolites from Gut Microbiome.BMC Genomics. 2020 Jul 20;21(Suppl 4):256. doi: 10.1186/s12864-020-6652-7. BMC Genomics. 2020. PMID: 32689932 Free PMC article.
-
Gut microbiome-metabolome interactions predict host condition.Microbiome. 2024 Feb 10;12(1):24. doi: 10.1186/s40168-023-01737-1. Microbiome. 2024. PMID: 38336867 Free PMC article.
-
A decrease in iron availability to human gut microbiome reduces the growth of potentially pathogenic gut bacteria; an in vitro colonic fermentation study.J Nutr Biochem. 2019 May;67:20-27. doi: 10.1016/j.jnutbio.2019.01.010. Epub 2019 Jan 27. J Nutr Biochem. 2019. PMID: 30831460 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
