

doi: 10.1186/s13059-015-0758-2.
An ensemble approach to accurately detect somatic mutations using SomaticSeq
Affiliations
- PMID: 26381235
- PMCID: PMC4574535
- DOI: 10.1186/s13059-015-0758-2
Item in Clipboard
An ensemble approach to accurately detect somatic mutations using SomaticSeq
Genome Biol.
.
Abstract
SomaticSeq is an accurate somatic mutation detection pipeline implementing a stochastic boosting algorithm to produce highly accurate somatic mutation calls for both single nucleotide variants and small insertions and deletions. The workflow currently incorporates five state-of-the-art somatic mutation callers, and extracts over 70 individual genomic and sequencing features for each candidate site. A training set is provided to an adaptively boosted decision tree learner to create a classifier for predicting mutation statuses. We validate our results with both synthetic and real data. We report that SomaticSeq is able to achieve better overall accuracy than any individual tool incorporated.
Figures
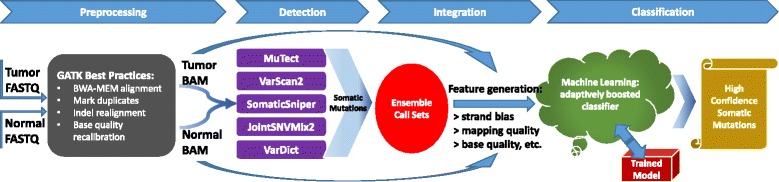
SomaticSeq workflow. The workflow starts with FASTQ files for both the tumor and the matched normal sequencing reads, which are processed using Genome Analysis Toolkit (GATK) best practices to create two BAM files. The five somatic SNV callers (and three indel callers) are run on the pair of BAM files to generate mutation calls. Their results are merged, and then up to 72 features for each of the combined calls are generated from the BAM files using SAMtools and GATK HaplotypeCaller, as well as outputs from the callers themselves. The ensemble along with the feature set is then provided to the machine-learning model, which is trained with either a separate data set or a portion of these data. After training, the model calculates the probability for each call, yielding a high-confidence somatic mutation call set
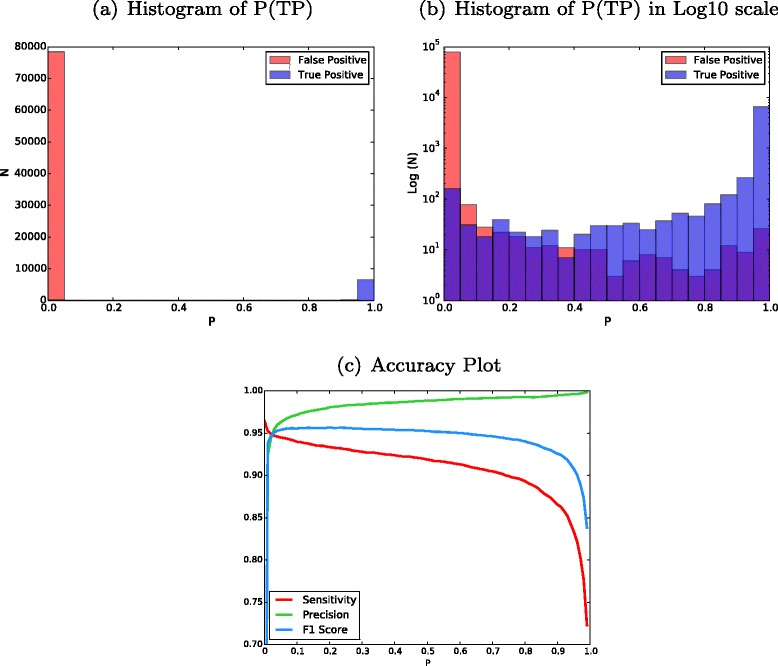
DREAM Challenge Stage 3 results trained from modified Stage 2 data. a Histogram of probability values (P) of all the mutation candidates in Stage 3. Higher probability values (closer to 1) imply that calls are more likely true somatic mutations. b The same plot with the y-axis in log10 scale. The overlaps can be seen. Keep in mind each unit in the y-axis is a tenfold increase. c An accuracy plot showing sensitivity, precision, and F
1 scores vs. P cut-off
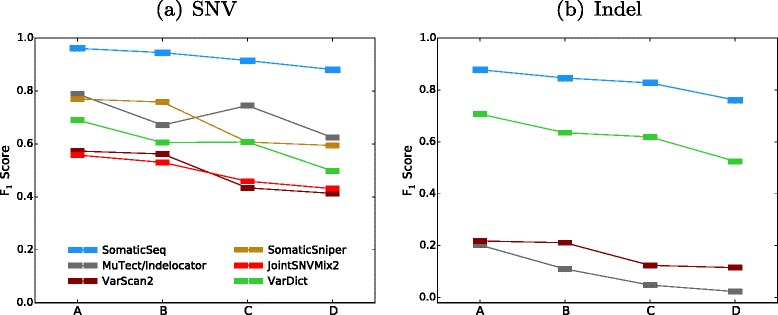
F
1 scores of SomaticSeq and the individual tools for the DREAM Challenge Stage 3 cross-validation. On the x-axes, Setting A is the pure normal/pure tumor. Setting B is the contaminated normal/pure tumor. Setting C is the pure normal/contaminated tumor. Setting D is the contaminated normal/contaminated tumor. a SNV results. b Indel results
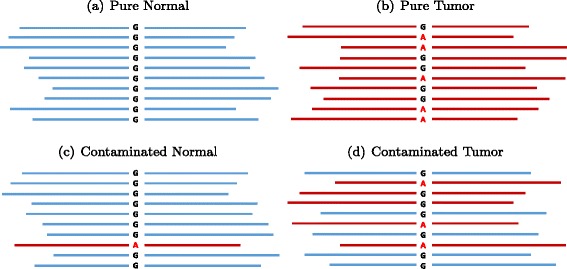
In silico titration of two human genomes. Blue represents reads from NA12878 (designated normal). Red represents reads from NS12911 (designated tumor). Going from (a) to (b) represents a somatic mutation of G >A, where G in the normal is a homozygous reference and A in the tumor is a heterozygous variant. c A normal contaminated with tumor tissues. d A tumor sample contaminated with normal tissues
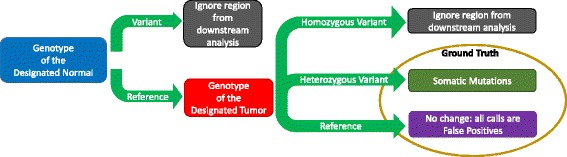
Obtaining the ground truth for the in silico tumor–normal data. In the NA12878 and NS12911 mixture, there are a total of 746,280 virtual somatic SNVs and 64,399 virtual somatic indels. A total of 2.2 billion high-confidence sites are interrogated (the remaining are ignored). During our analyses, a somatic mutation rate of one out of a million was enforced to represent a realistic prior probability of somatic mutations
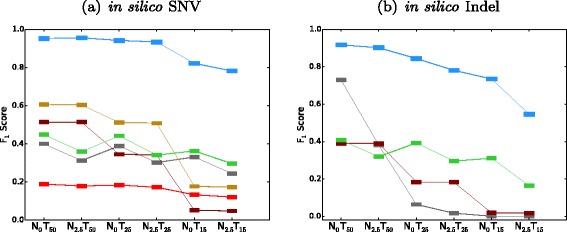
F
1 scores of SomaticSeq and the individual tools in in silico titration. Color legends are shown in Fig. 3. On the x-axes, the subscript denotes the expected VAF as a percentage, i.e., N 0
T
50 means the normal has VAF = 0 % (i.e., pure normal) and the tumor has VAF = 50 %. N 2.5
T
15 represents a challenging data set where VAF = 2.5 % for normal and VAF = 15 % for tumor, i.e., 5 % of the normal sample is contaminated with tumor tissues and 30 % of the tumor sample is contaminated with normal tissues. a SNV results. b Indel results
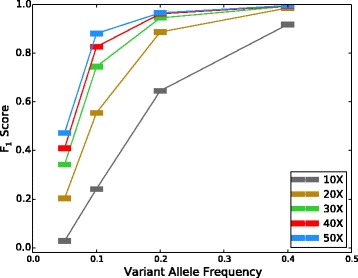
F
1 scores vs. VAF for different coverage depths
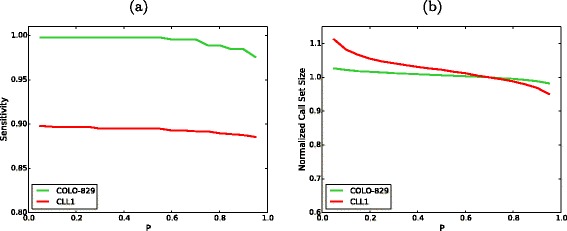
SomaticSeq performance on real data. a The sensitivity of SomaticSeq as a function of P cut-offs. b The call set size as a function of P cut-offs, normalized to the call set size at P=0.7, i.e., the ratio between the call set size at a given P and the call set size at P=0.7 (default cut-off in this study)
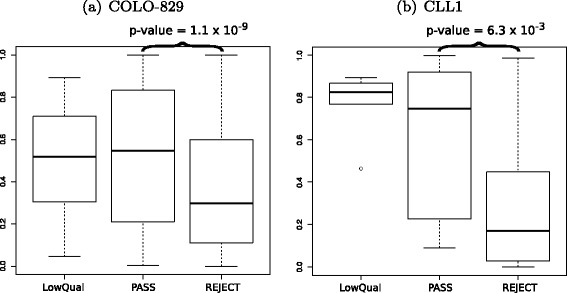
CADD scores from SomaticSeq’s PASS (high confidence) calls vs. LowQual (medium confidence) vs. REJECT (likely false positive) calls. a COLO-829. b CLL1. Only non-synonymous SNVs were evaluated. The p-values were calculated from a two-sided Wilcoxon rank-sum test
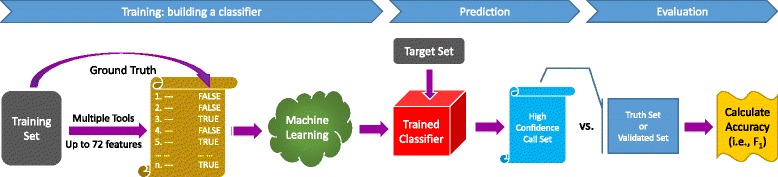
Machine learning: a training set with ground truth is provided to the machine-learning algorithm to create an adaptively boosted classifier. The classifier is applied to a target set to create a high-confidence somatic mutation call set. The call set is compared to the ground truth or the validated mutation list of the target set to calculate the accuracy (only sensitivity is calculated for real data)
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
