

Tools and best practices for data processing in allelic expression analysis
- PMID: 26381377
- PMCID: PMC4574606
- DOI: 10.1186/s13059-015-0762-6
Tools and best practices for data processing in allelic expression analysis
Abstract
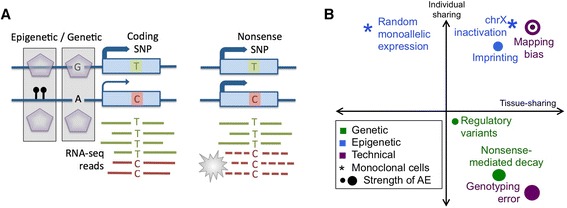
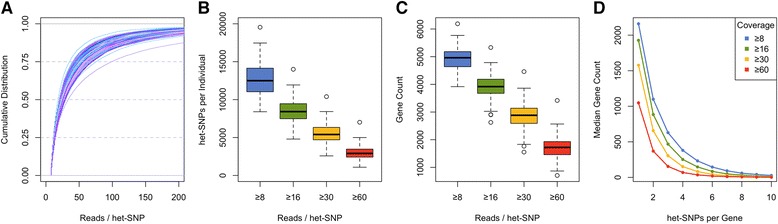
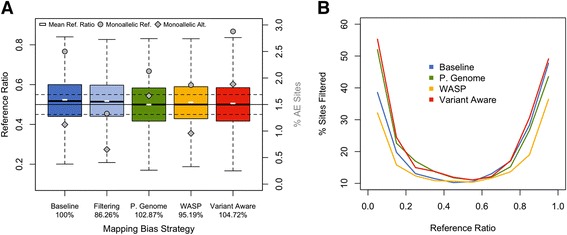
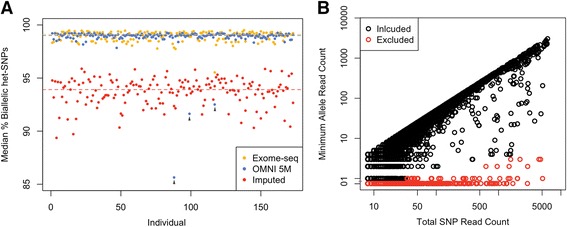
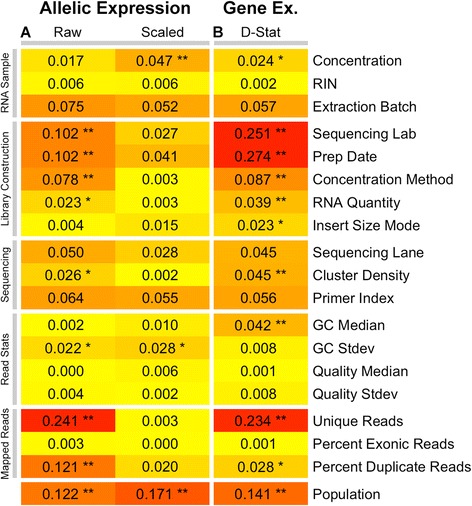
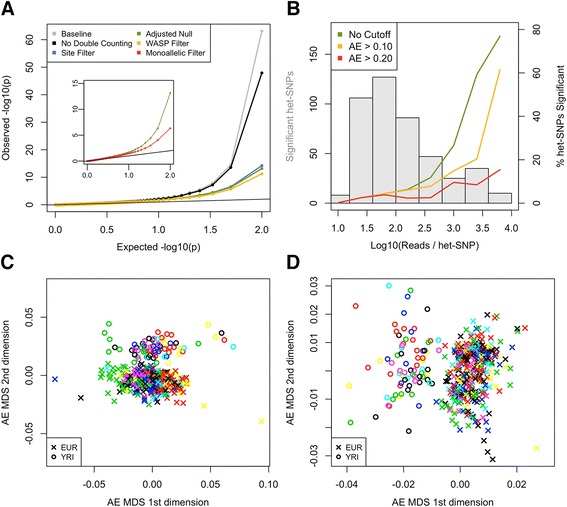
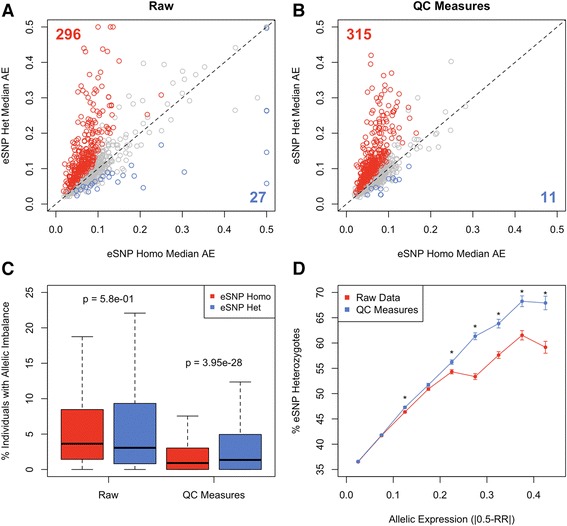
Allelic expression analysis has become important for integrating genome and transcriptome data to characterize various biological phenomena such as cis-regulatory variation and nonsense-mediated decay. We analyze the properties of allelic expression read count data and technical sources of error, such as low-quality or double-counted RNA-seq reads, genotyping errors, allelic mapping bias, and technical covariates due to sample preparation and sequencing, and variation in total read depth. We provide guidelines for correcting such errors, show that our quality control measures improve the detection of relevant allelic expression, and introduce tools for the high-throughput production of allelic expression data from RNA-sequencing data.
Figures
References
Publication types
MeSH terms
Grants and funding
- R01 DA006227/DA/NIDA NIH HHS/United States
- U01 HG006569/HG/NHGRI NIH HHS/United States
- R01 MH090936/MH/NIMH NIH HHS/United States
- 3R01MH101814-02S1/MH/NIMH NIH HHS/United States
- R01 MH090951/MH/NIMH NIH HHS/United States
- 5U01HG006569/HG/NHGRI NIH HHS/United States
- R01 MH090948/MH/NIMH NIH HHS/United States
- R01 MH090941/MH/NIMH NIH HHS/United States
- HHSN261200800001C/RC/CCR NIH HHS/United States
- R01 MH090937/MH/NIMH NIH HHS/United States
- HHSN268201000029C/HL/NHLBI NIH HHS/United States
- HHSN261200800001E/CA/NCI NIH HHS/United States
- R01 MH101814/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
