

MYC, Metabolism, and Cancer
- PMID: 26382145
- PMCID: PMC4592441
- DOI: 10.1158/2159-8290.CD-15-0507
MYC, Metabolism, and Cancer
Abstract
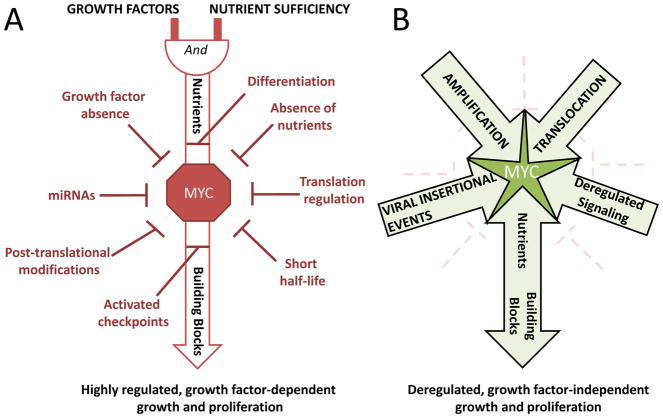
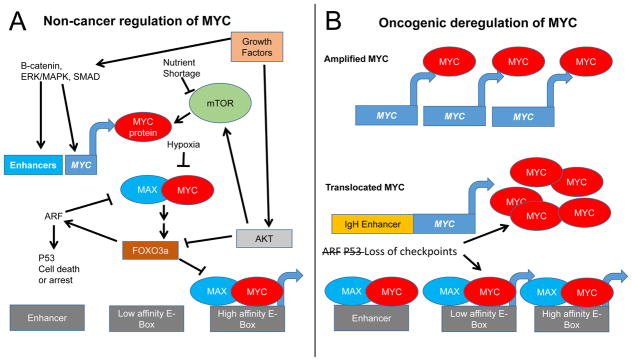
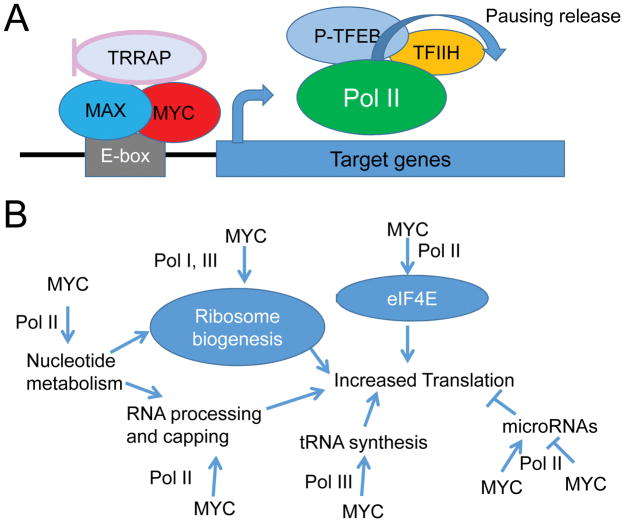
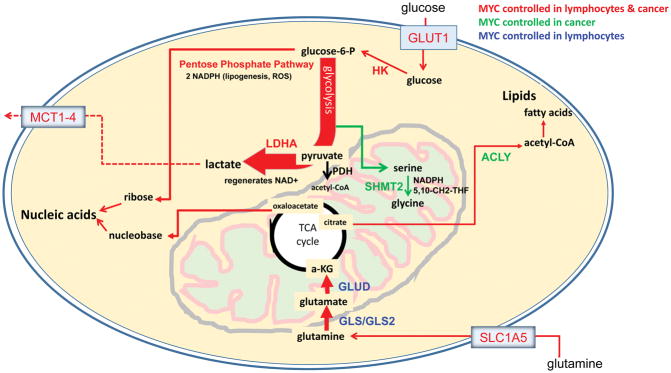
The MYC oncogene encodes a transcription factor, MYC, whose broad effects make its precise oncogenic role enigmatically elusive. The evidence to date suggests that MYC triggers selective gene expression amplification to promote cell growth and proliferation. Through its targets, MYC coordinates nutrient acquisition to produce ATP and key cellular building blocks that increase cell mass and trigger DNA replication and cell division. In cancer, genetic and epigenetic derangements silence checkpoints and unleash MYC's cell growth- and proliferation-promoting metabolic activities. Unbridled growth in response to deregulated MYC expression creates dependence on MYC-driven metabolic pathways, such that reliance on specific metabolic enzymes provides novel targets for cancer therapy.
Significance: MYC's expression and activity are tightly regulated in normal cells by multiple mechanisms, including a dependence upon growth factor stimulation and replete nutrient status. In cancer, genetic deregulation of MYC expression and loss of checkpoint components, such as TP53, permit MYC to drive malignant transformation. However, because of the reliance of MYC-driven cancers on specific metabolic pathways, synthetic lethal interactions between MYC overexpression and specific enzyme inhibitors provide novel cancer therapeutic opportunities.
©2015 American Association for Cancer Research.
Conflict of interest statement
The authors declare no conflict of interest.
No potential conflicts of interest were disclosed.
Figures
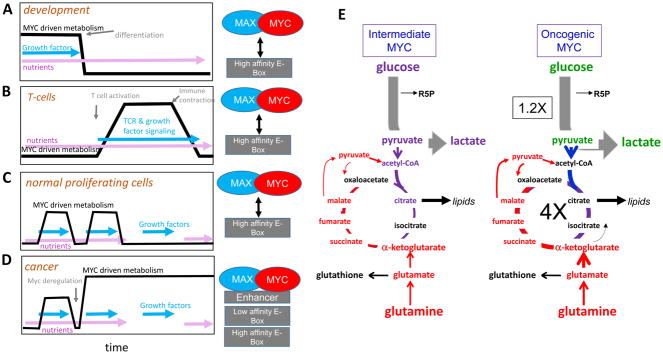
References
-
- Bister K, Jansen HW. Oncogenes in retroviruses and cells: biochemistry and molecular genetics. Advances in cancer research. 1986;47:99–188. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30CA16620/CA/NCI NIH HHS/United States
- R01 CA057341/CA/NCI NIH HHS/United States
- F32 CA174148/CA/NCI NIH HHS/United States
- T32 CA009140/CA/NCI NIH HHS/United States
- 5F32CA174148/CA/NCI NIH HHS/United States
- R01 CA051497/CA/NCI NIH HHS/United States
- F30CA200347/CA/NCI NIH HHS/United States
- 1F32CA180370/CA/NCI NIH HHS/United States
- P30 CA016520/CA/NCI NIH HHS/United States
- F32 CA180370/CA/NCI NIH HHS/United States
- R01CA057341/CA/NCI NIH HHS/United States
- R01CA051497/CA/NCI NIH HHS/United States
- 5T32CA009140-40/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
