

Serum Albumin Domain Structures in Human Blood Serum by Mass Spectrometry and Computational Biology
- PMID: 26385339
- PMCID: PMC4813692
- DOI: 10.1074/mcp.M115.048504
Serum Albumin Domain Structures in Human Blood Serum by Mass Spectrometry and Computational Biology
Abstract
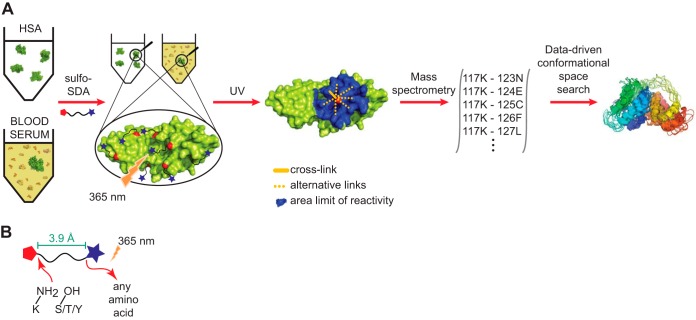
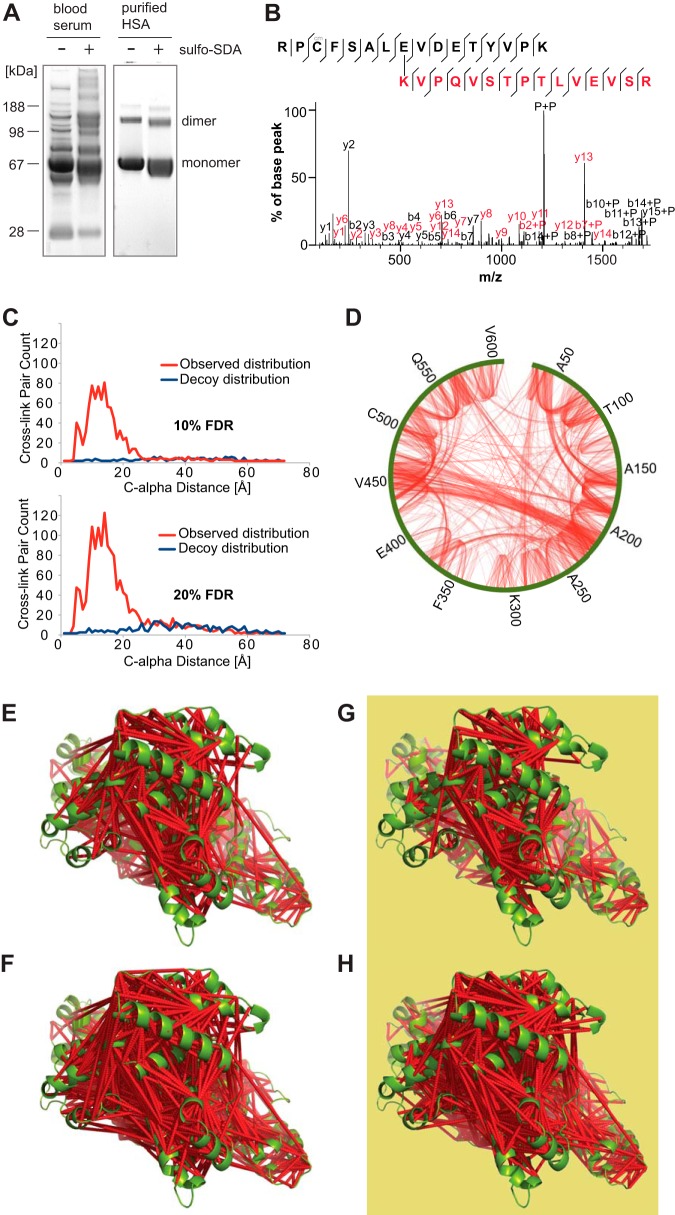
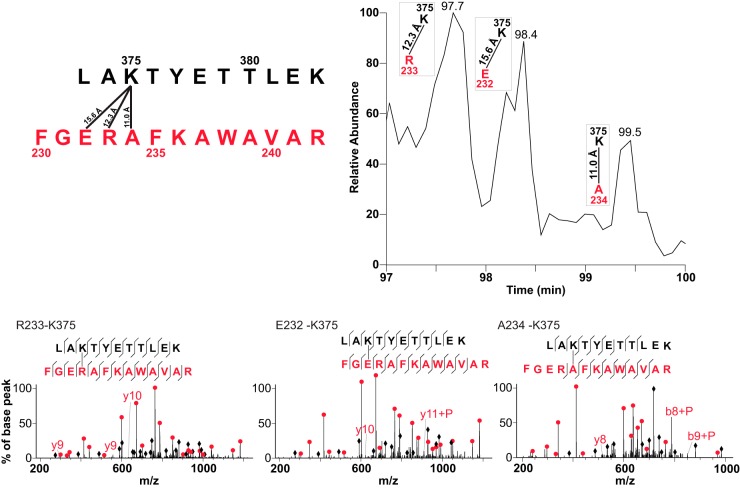
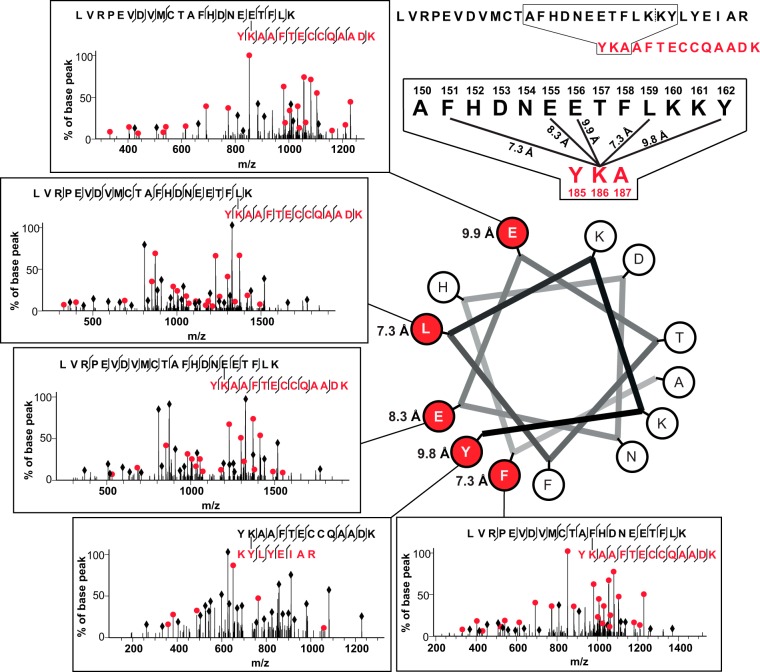
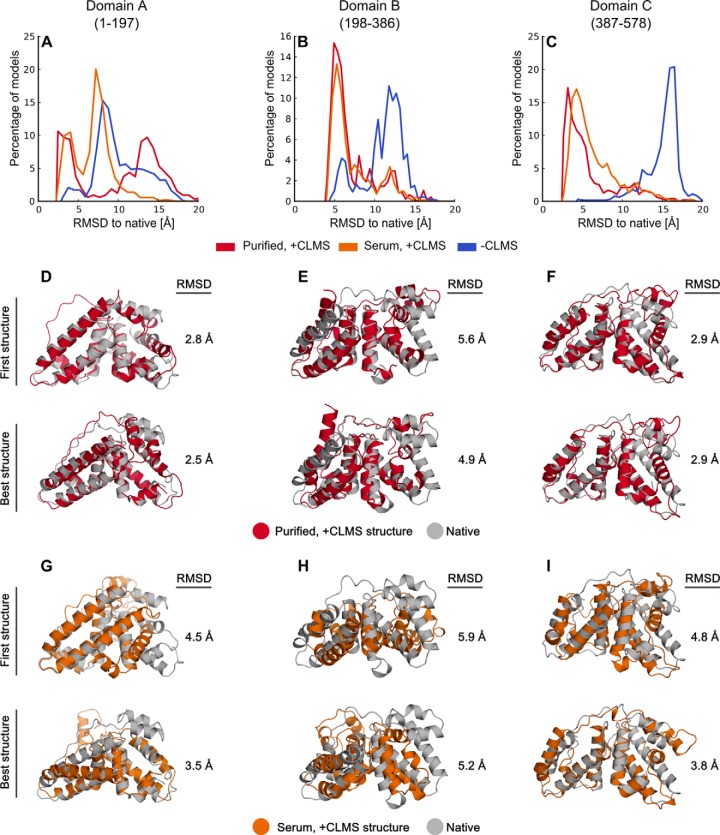
Chemical cross-linking combined with mass spectrometry has proven useful for studying protein-protein interactions and protein structure, however the low density of cross-link data has so far precluded its use in determining structures de novo. Cross-linking density has been typically limited by the chemical selectivity of the standard cross-linking reagents that are commonly used for protein cross-linking. We have implemented the use of a heterobifunctional cross-linking reagent, sulfosuccinimidyl 4,4'-azipentanoate (sulfo-SDA), combining a traditional sulfo-N-hydroxysuccinimide (sulfo-NHS) ester and a UV photoactivatable diazirine group. This diazirine yields a highly reactive and promiscuous carbene species, the net result being a greatly increased number of cross-links compared with homobifunctional, NHS-based cross-linkers. We present a novel methodology that combines the use of this high density photo-cross-linking data with conformational space search to investigate the structure of human serum albumin domains, from purified samples, and in its native environment, human blood serum. Our approach is able to determine human serum albumin domain structures with good accuracy: root-mean-square deviation to crystal structure are 2.8/5.6/2.9 Å (purified samples) and 4.5/5.9/4.8Å (serum samples) for domains A/B/C for the first selected structure; 2.5/4.9/2.9 Å (purified samples) and 3.5/5.2/3.8 Å (serum samples) for the best out of top five selected structures. Our proof-of-concept study on human serum albumin demonstrates initial potential of our approach for determining the structures of more proteins in the complex biological contexts in which they function and which they may require for correct folding. Data are available via ProteomeXchange with identifier PXD001692.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Caffrey M. (2003) Membrane protein crystallization. J. Struct. Biol. 142, 108–132 - PubMed
-
- Rappsilber J., Siniossoglou S., Hurt E. C., and Mann M. (1999) A generic strategy to analyze the spatial organization of multi-protein complexes by cross-linking and mass spectrometry. Anal. Chem. 72, 267–275 - PubMed
-
- Boutet S., Lomb L., Williams G. J., Barends T. R. M., Aquila A., Doak R. B., Weierstall U., DePonte D. P., Steinbrener J., Shoeman R. L., Messerschmidt M., Barty A., White T. A., Kassemeyer S., Kirian R. A., Seibert M. M., Montanez P. A., Kenney C., Herbst R., Hart P., Pines J., Haller G., Gruner S. M., Philipp H. T., Tate M. W., Hromalik M., Koerner L. J., van Bakel N., Morse J., Ghonsalves W., Arnlund D., Bogan M. J., Caleman C., Fromme R., Hampton C. Y., Hunter M. S., Johansson L. C., Katona G., Kupitz C., Liang M., Martin A. V., Nass K., Redecke L., Stellato F., Timneanu N., Wang D., Zatsepin N. A., Schafer D., Defever J., Neutze R., Fromme P., Spence J. C. H., Chapman H. N., and Schlichting I. (2012) High-resolution protein structure determination by serial femtosecond crystallography. Science 337, 362–364 - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
