

Ultrastructural studies of ALS mitochondria connect altered function and permeability with defects of mitophagy and mitochondriogenesis
- PMID: 26388731
- PMCID: PMC4555074
- DOI: 10.3389/fncel.2015.00341
Ultrastructural studies of ALS mitochondria connect altered function and permeability with defects of mitophagy and mitochondriogenesis
Abstract
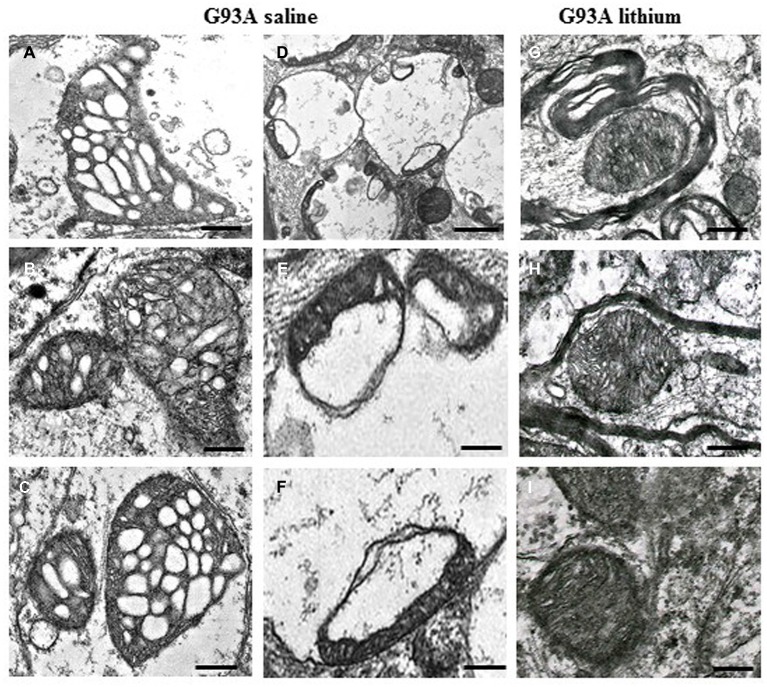
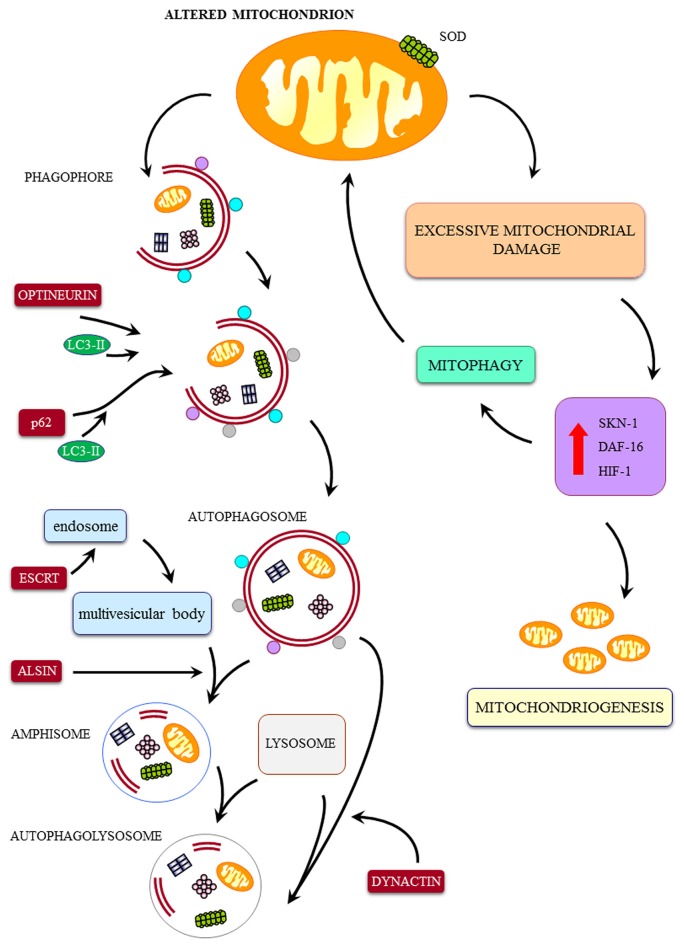
The key role of mitochondria in patients affected by amyotrophic lateral sclerosis (ALS) is well documented by electron microscopy studies of motor neurons within spinal cord and brainstem. Nonetheless, recent studies challenged the role of mitochondria placed within the cell body of motor neuron. In fact, it was demonstrated that, despite preservation of mitochondria placed within this compartment, there is no increase in the lifespan of transgenic mouse models of ALS. Thus, the present mini-review comments on morphological findings of mitochondrial alterations in ALS patients in connection with novel findings about mitochondrial dynamics within various compartments of motor neurons. The latter issue was recently investigated in relationship with altered calcium homeostasis and autophagy, which affect mitochondria in ALS. In fact, it was recently indicated that a pathological mitophagy, mitochondriogenesis and calcium homeostasis produce different ultrastructural effects within specific regions of motor neurons. This might explain why specific compartments of motor neurons possess different thresholds to mitochondrial damage. In particular, it appears that motor axons represent the most sensitive compartment which undergoes the earliest and most severe alterations in the course of ALS. It is now evident that altered calcium buffering is compartment-dependent, as well as mitophagy and mitochondriogenesis. On the other hand, mitochondrial homeostasis strongly relies on calcium handling, the removal of altered mitochondria through the autophagy flux (mitophagy) and the biogenesis of novel mitochondria (mitochondriogenesis). Thus, recent findings related to altered calcium storage and impaired autophagy flux in ALS may help to understand the occurrence of mitochondrial alterations as a hallmark in ALS patients. At the same time, the compartmentalization of such dysfunctions may be explained considering the compartments of calcium dynamics and autophagy flux within motor neurons.
Keywords: amyotrophic lateral sclerosis; autophagy; biogenesis of mitochondria; electron microscopy; human patients; mitochondria; motor neuron.
Figures
Similar articles
-
Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis.Front Cell Neurosci. 2015 Nov 6;9:434. doi: 10.3389/fncel.2015.00434. eCollection 2015. Front Cell Neurosci. 2015. PMID: 26594150 Free PMC article.
-
Mitochondrial dynamic abnormalities in amyotrophic lateral sclerosis.Transl Neurodegener. 2015 Jul 29;4:14. doi: 10.1186/s40035-015-0037-x. eCollection 2015. Transl Neurodegener. 2015. PMID: 26225210 Free PMC article.
-
The role of insulin-like growth factor 1 in ALS cell and mouse models: A mitochondrial protector.Brain Res Bull. 2019 Jan;144:1-13. doi: 10.1016/j.brainresbull.2018.09.015. Epub 2018 Nov 8. Brain Res Bull. 2019. PMID: 30414993
-
Exploring new pathways of neurodegeneration in ALS: the role of mitochondria quality control.Brain Res. 2015 May 14;1607:36-46. doi: 10.1016/j.brainres.2014.09.065. Epub 2014 Oct 6. Brain Res. 2015. PMID: 25301687 Free PMC article. Review.
-
Mitochondrial involvement in amyotrophic lateral sclerosis.Neurochem Int. 2002 May;40(6):543-51. doi: 10.1016/s0197-0186(01)00125-5. Neurochem Int. 2002. PMID: 11850111 Review.
Cited by
-
Quantitative Ultrastructural Morphometry and Gene Expression of mTOR-Related Mitochondriogenesis within Glioblastoma Cells.Int J Mol Sci. 2020 Jun 27;21(13):4570. doi: 10.3390/ijms21134570. Int J Mol Sci. 2020. PMID: 32604996 Free PMC article.
-
Amyotrophic lateral sclerosis, gene deregulation in the anterior horn of the spinal cord and frontal cortex area 8: implications in frontotemporal lobar degeneration.Aging (Albany NY). 2017 Mar 9;9(3):823-851. doi: 10.18632/aging.101195. Aging (Albany NY). 2017. PMID: 28283675 Free PMC article.
-
Impaired Mitophagy Plays a Role in Denervation of Neuromuscular Junctions in ALS Mice.Front Neurosci. 2017 Aug 25;11:473. doi: 10.3389/fnins.2017.00473. eCollection 2017. Front Neurosci. 2017. PMID: 28890682 Free PMC article.
-
Skeletal Muscle in ALS: An Unappreciated Therapeutic Opportunity?Cells. 2021 Mar 2;10(3):525. doi: 10.3390/cells10030525. Cells. 2021. PMID: 33801336 Free PMC article. Review.
-
FUS Mutation Causes Disordered Lipid Metabolism in Skeletal Muscle Associated with ALS.Mol Neurobiol. 2022 Dec;59(12):7265-7277. doi: 10.1007/s12035-022-03048-2. Epub 2022 Sep 28. Mol Neurobiol. 2022. PMID: 36169888
References
-
- Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., et al. . (2006). TDP-43 is a component of ubiquitin-positive tau negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. 10.1016/j.bbrc.2006.10.093 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous