

Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells
- PMID: 26389676
- PMCID: PMC4607115
- DOI: 10.1172/JCI82498
Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells
Abstract
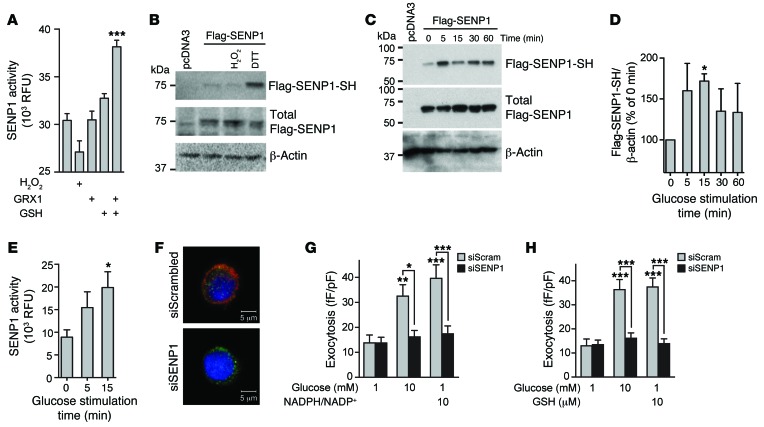
Insulin secretion from β cells of the pancreatic islets of Langerhans controls metabolic homeostasis and is impaired in individuals with type 2 diabetes (T2D). Increases in blood glucose trigger insulin release by closing ATP-sensitive K+ channels, depolarizing β cells, and opening voltage-dependent Ca2+ channels to elicit insulin exocytosis. However, one or more additional pathway(s) amplify the secretory response, likely at the distal exocytotic site. The mitochondrial export of isocitrate and engagement with cytosolic isocitrate dehydrogenase (ICDc) may be one key pathway, but the mechanism linking this to insulin secretion and its role in T2D have not been defined. Here, we show that the ICDc-dependent generation of NADPH and subsequent glutathione (GSH) reduction contribute to the amplification of insulin exocytosis via sentrin/SUMO-specific protease-1 (SENP1). In human T2D and an in vitro model of human islet dysfunction, the glucose-dependent amplification of exocytosis was impaired and could be rescued by introduction of signaling intermediates from this pathway. Moreover, islet-specific Senp1 deletion in mice caused impaired glucose tolerance by reducing the amplification of insulin exocytosis. Together, our results identify a pathway that links glucose metabolism to the amplification of insulin secretion and demonstrate that restoration of this axis rescues β cell function in T2D.
Figures







Comment in
-
How do reducing equivalents increase insulin secretion?J Clin Invest. 2015 Oct 1;125(10):3754-6. doi: 10.1172/JCI84011. Epub 2015 Sep 21. J Clin Invest. 2015. PMID: 26389672 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
