

Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma
- PMID: 26390243
- PMCID: PMC4959788
- DOI: 10.1038/nm.3952
Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma
Erratum in
-
Author Correction: Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma.Nat Med. 2024 Jul;30(7):2090. doi: 10.1038/s41591-024-03054-y. Nat Med. 2024. PMID: 38816611 No abstract available.
Abstract
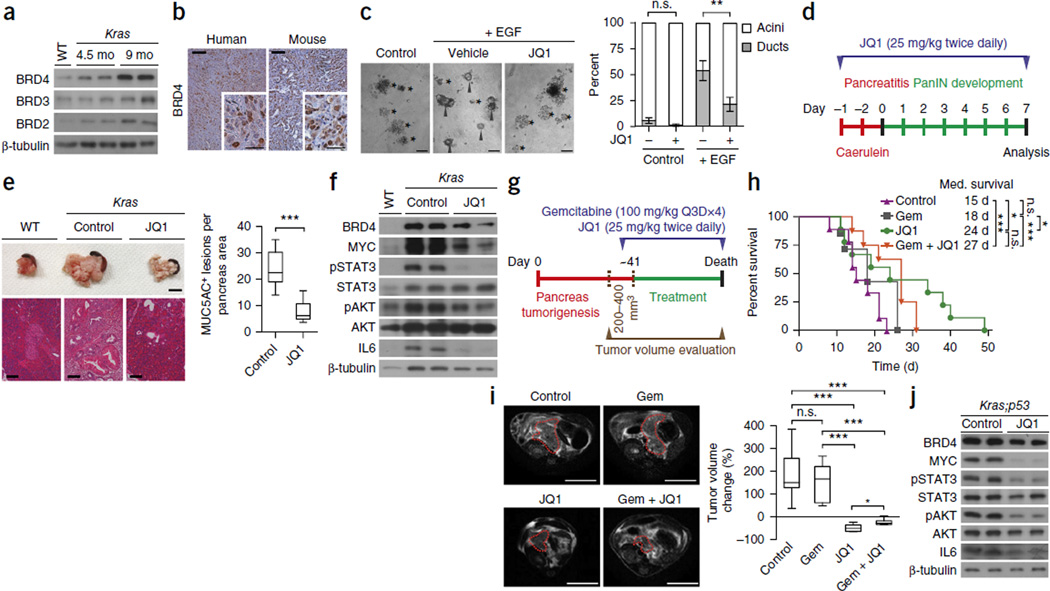
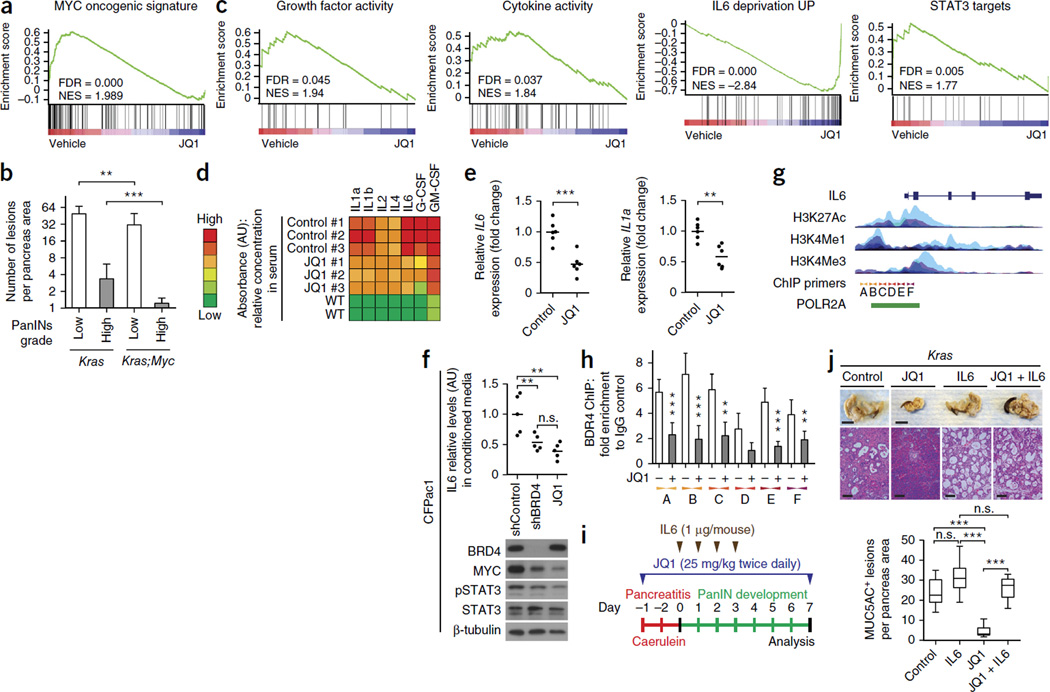
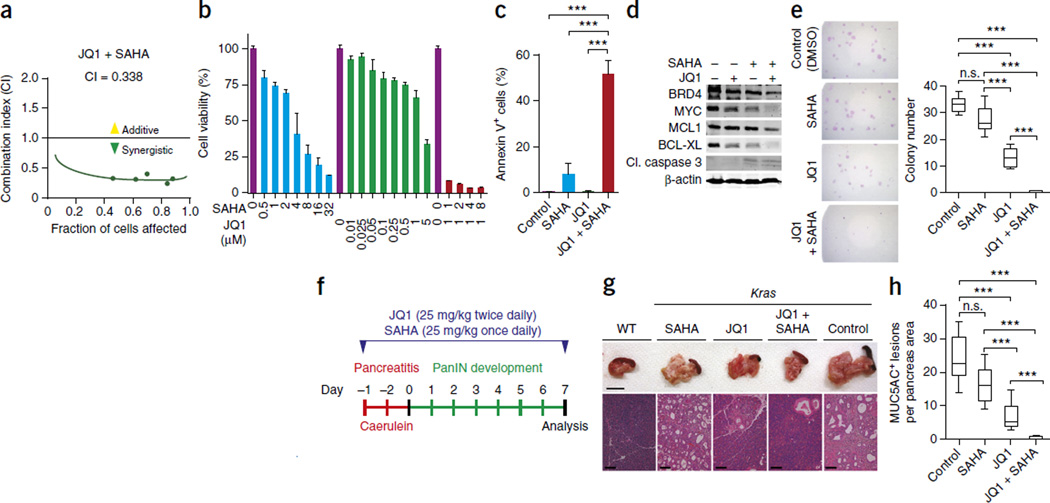
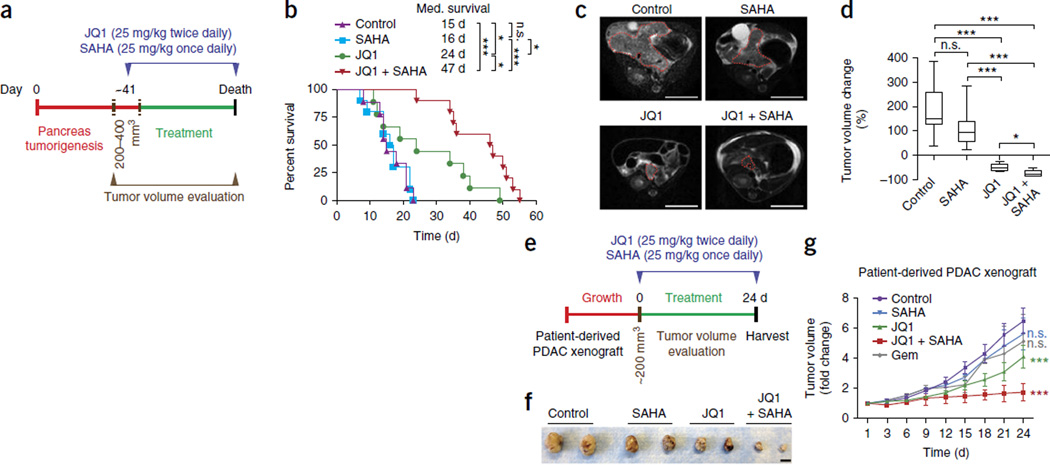
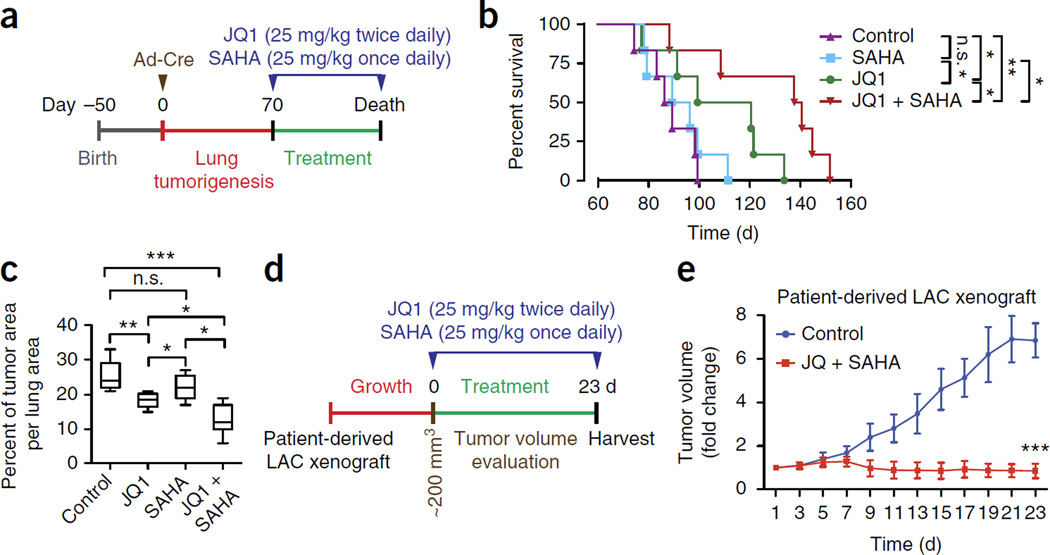
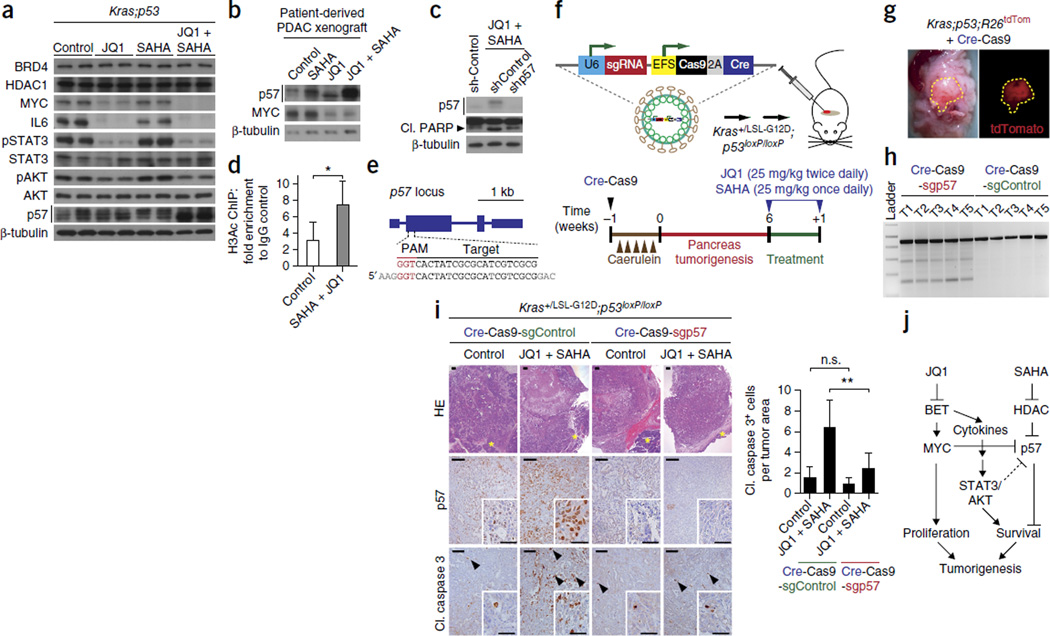
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal human cancers and shows resistance to any therapeutic strategy used. Here we tested small-molecule inhibitors targeting chromatin regulators as possible therapeutic agents in PDAC. We show that JQ1, an inhibitor of the bromodomain and extraterminal (BET) family of proteins, suppresses PDAC development in mice by inhibiting both MYC activity and inflammatory signals. The histone deacetylase (HDAC) inhibitor SAHA synergizes with JQ1 to augment cell death and more potently suppress advanced PDAC. Finally, using a CRISPR-Cas9-based method for gene editing directly in the mouse adult pancreas, we show that de-repression of p57 (also known as KIP2 or CDKN1C) upon combined BET and HDAC inhibition is required for the induction of combination therapy-induced cell death in PDAC. SAHA is approved for human use, and molecules similar to JQ1 are being tested in clinical trials. Thus, these studies identify a promising epigenetic-based therapeutic strategy that may be rapidly implemented in fatal human tumors.
Figures
Comment in
-
Therapy: Targeting chromatin remodelling proteins to treat pancreatic cancer.Nat Rev Gastroenterol Hepatol. 2015 Nov;12(11):608. doi: 10.1038/nrgastro.2015.171. Epub 2015 Oct 6. Nat Rev Gastroenterol Hepatol. 2015. PMID: 26441249 No abstract available.
References
-
- Rahib L, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. - PubMed
-
- Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. - PubMed
-
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014;371:1039–1049. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
