

Role of T-cell reconstitution in HIV-1 antiretroviral therapy-induced bone loss
- PMID: 26392000
- PMCID: PMC4580984
- DOI: 10.1038/ncomms9282
Role of T-cell reconstitution in HIV-1 antiretroviral therapy-induced bone loss
Abstract
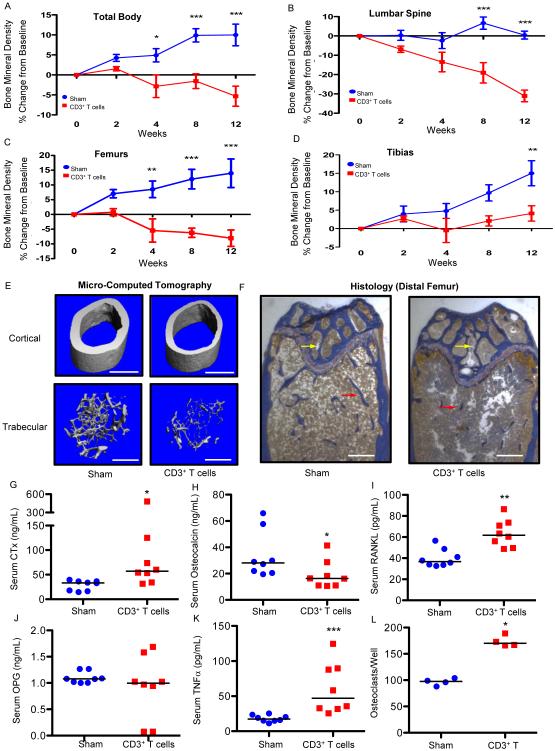
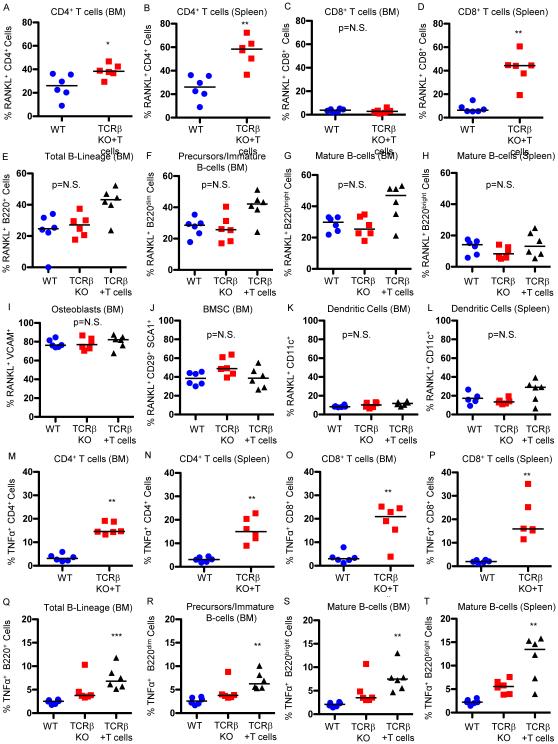
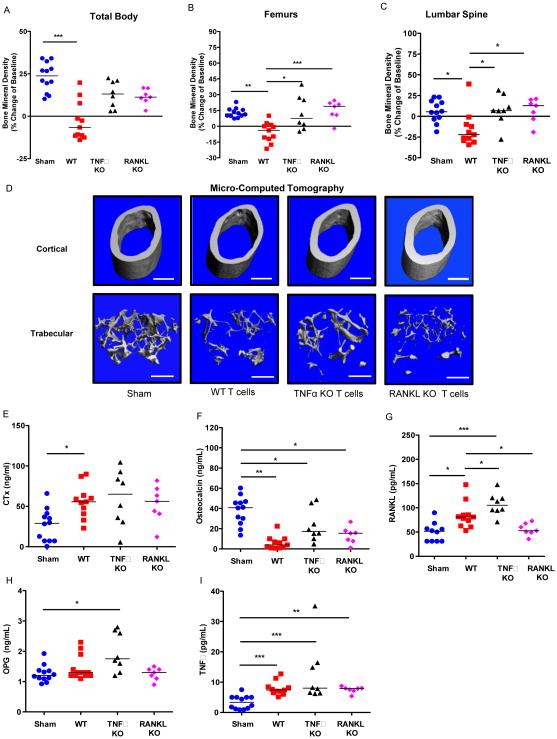
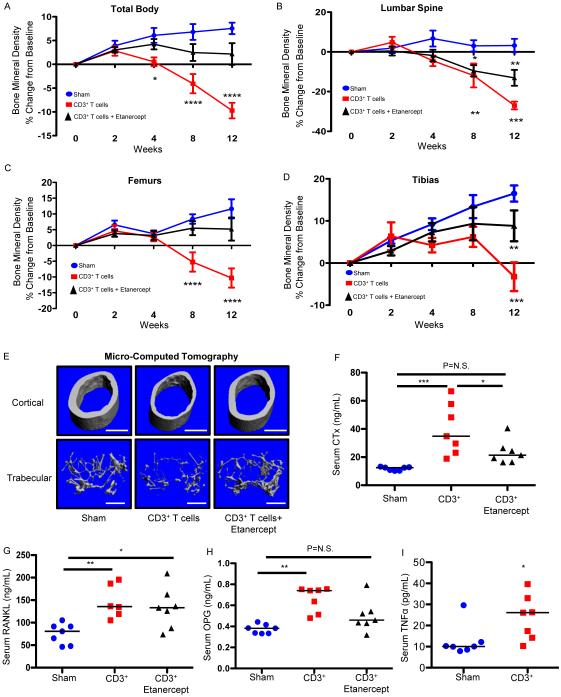
HIV infection causes bone loss. We previously reported that immunosuppression-mediated B-cell production of receptor activator of NF-κB ligand (RANKL) coupled with decline in osteoprotegerin correlate with decreased bone mineral density (BMD) in untreated HIV infection. Paradoxically, antiretroviral therapy (ART) worsens bone loss although existing data suggest that such loss is largely independent of specific antiretroviral regimen. This led us to hypothesize that skeletal deterioration following HIV disease reversal with ART may be related to T-cell repopulation and/or immune reconstitution. Here we transplant T cells into immunocompromised mice to mimic ART-induced T-cell expansion. T-cell reconstitution elicits RANKL and TNFα production by B cells and/or T cells, accompanied by enhanced bone resorption and BMD loss. Reconstitution of TNFα- or RANKL-null T-cells and pharmacological TNFα antagonist all protect cortical, but not trabecular bone, revealing complex effects of T-cell reconstitution on bone turnover. These findings suggest T-cell repopulation and/or immune reconstitution as putative mechanisms for bone loss following ART initiation.
Figures
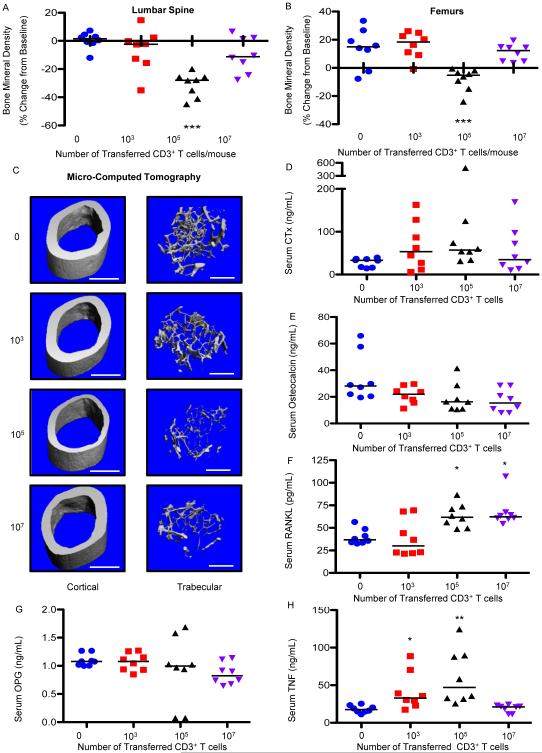
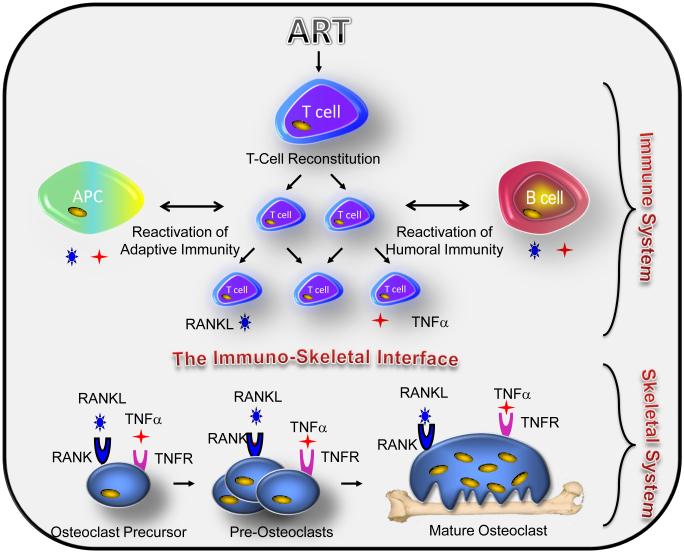
Comment in
-
Inflammation: Immune recovery drives bone loss.Nat Rev Endocrinol. 2015 Dec;11(12):689. doi: 10.1038/nrendo.2015.180. Epub 2015 Oct 13. Nat Rev Endocrinol. 2015. PMID: 26460343 No abstract available.
References
-
- Thomas J, Doherty SM. HIV infection--a risk factor for osteoporosis. J Acquir Immune Defic Syndr. 2003;33:281–291. - PubMed
-
- Stone B, Dockrell D, Bowman C, McCloskey E. HIV and bone disease. Arch Biochem Biophys. 2010;503:66–77. - PubMed
-
- Arora S, et al. HIV and bone loss. Curr Osteoporos Rep. 2010;8:219–226. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R38 AI140299/AI/NIAID NIH HHS/United States
- P30 AR046031/AR/NIAMS NIH HHS/United States
- R01AG040013/AG/NIA NIH HHS/United States
- P30AR46031/AR/NIAMS NIH HHS/United States
- P01 CA098912/CA/NCI NIH HHS/United States
- R01 AR059364/AR/NIAMS NIH HHS/United States
- R01AR059364/AR/NIAMS NIH HHS/United States
- R01 AR056090/AR/NIAMS NIH HHS/United States
- U54 AG062334/AG/NIA NIH HHS/United States
- R01 AR053898/AR/NIAMS NIH HHS/United States
- I01 BX000105/BX/BLRD VA/United States
- R01AR056090/AR/NIAMS NIH HHS/United States
- R01 AG040013/AG/NIA NIH HHS/United States
- R01AR053898/AR/NIAMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
