

Regulation of constitutive and alternative mRNA splicing across the human transcriptome by PRPF8 is determined by 5' splice site strength
- PMID: 26392272
- PMCID: PMC4578845
- DOI: 10.1186/s13059-015-0749-3
Regulation of constitutive and alternative mRNA splicing across the human transcriptome by PRPF8 is determined by 5' splice site strength
Abstract
Background: Sequential assembly of the human spliceosome on RNA transcripts regulates splicing across the human transcriptome. The core spliceosome component PRPF8 is essential for spliceosome assembly through its participation in ribonucleoprotein (RNP) complexes for splice-site recognition, branch-point formation and catalysis. PRPF8 deficiency is linked to human diseases like retinitis pigmentosa or myeloid neoplasia, but its genome-wide effects on constitutive and alternative splicing remain unclear.
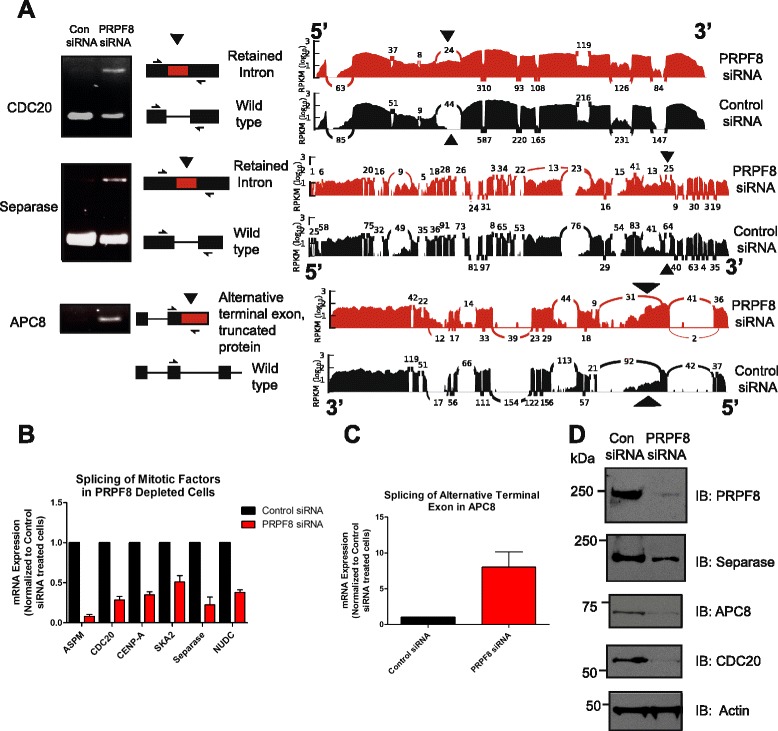
Results: Here, we show that alterations in RNA splicing patterns across the human transcriptome that occur in conditions of restricted cellular PRPF8 abundance are defined by the altered splicing of introns with weak 5' splice sites. iCLIP of spliceosome components reveals that PRPF8 depletion decreases RNP complex formation at most splice sites in exon-intron junctions throughout the genome. However, impaired splicing affects only a subset of human transcripts, enriched for mitotic cell cycle factors, leading to mitotic arrest. Preferentially retained introns and differentially used exons in the affected genes contain weak 5' splice sites, but are otherwise indistinguishable from adjacent spliced introns. Experimental enhancement of splice-site strength in mini-gene constructs overcomes the effects of PRPF8 depletion on the kinetics and fidelity of splicing during transcription.
Conclusions: Competition for PRPF8 availability alters the transcription-coupled splicing of RNAs in which weak 5' splice sites predominate, enabling diversification of human gene expression during biological processes like mitosis. Our findings exemplify the regulatory potential of changes in the core spliceosome machinery, which may be relevant to slow-onset human genetic diseases linked to PRPF8 deficiency.
Figures








References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
