

Bacterial infection remodels the DNA methylation landscape of human dendritic cells
- PMID: 26392366
- PMCID: PMC4665002
- DOI: 10.1101/gr.192005.115
Bacterial infection remodels the DNA methylation landscape of human dendritic cells
Abstract
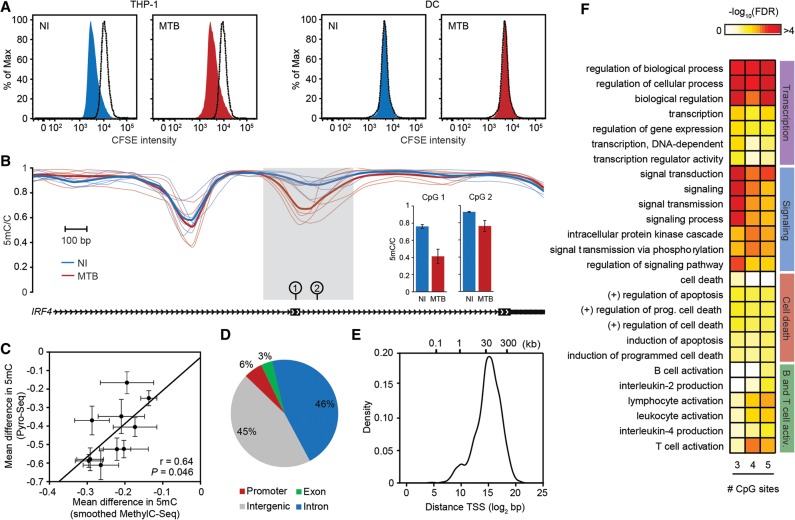
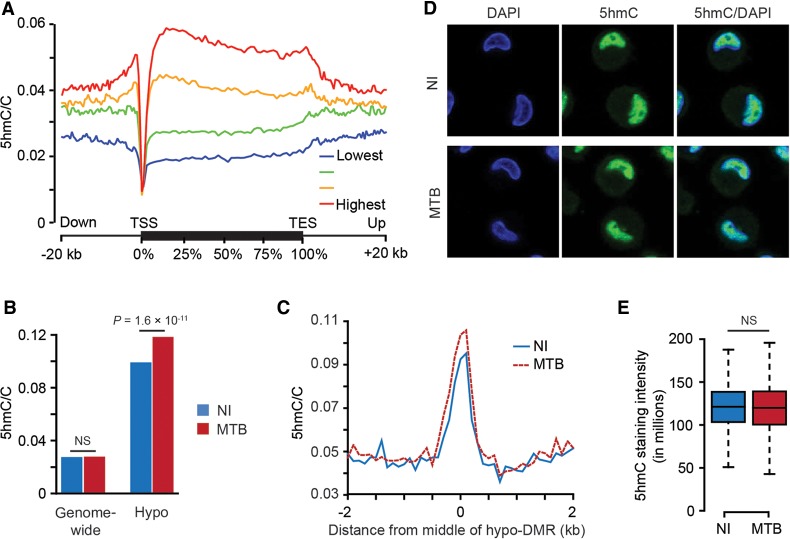
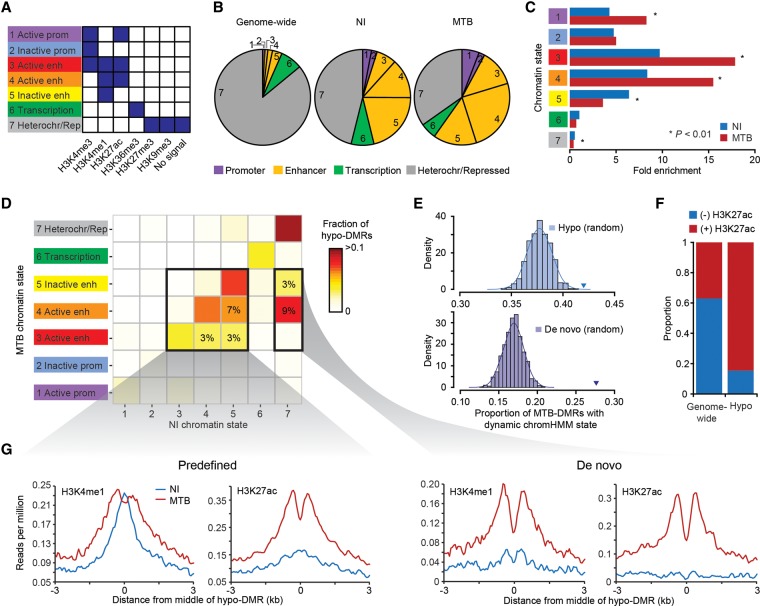
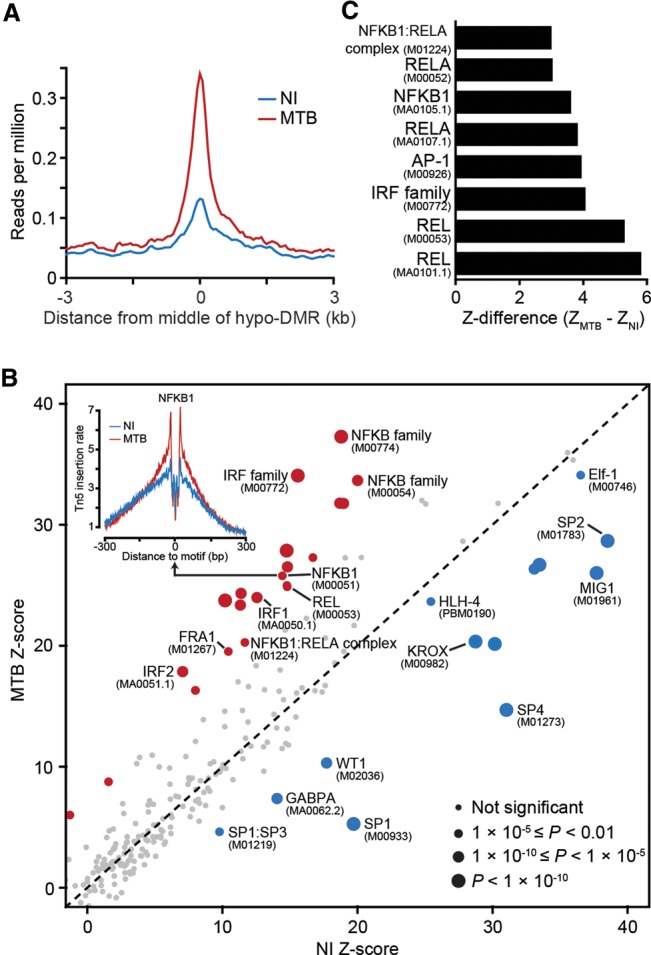
DNA methylation is an epigenetic mark thought to be robust to environmental perturbations on a short time scale. Here, we challenge that view by demonstrating that the infection of human dendritic cells (DCs) with a live pathogenic bacteria is associated with rapid and active demethylation at thousands of loci, independent of cell division. We performed an integrated analysis of data on genome-wide DNA methylation, histone mark patterns, chromatin accessibility, and gene expression, before and after infection. We found that infection-induced demethylation rarely occurs at promoter regions and instead localizes to distal enhancer elements, including those that regulate the activation of key immune transcription factors. Active demethylation is associated with extensive epigenetic remodeling, including the gain of histone activation marks and increased chromatin accessibility, and is strongly predictive of changes in the expression levels of nearby genes. Collectively, our observations show that active, rapid changes in DNA methylation in enhancers play a previously unappreciated role in regulating the transcriptional response to infection, even in nonproliferating cells.
© 2015 Pacis et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD. 2013. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc 8: 1765–1786. - PubMed
-
- Ardeshna KM, Pizzey AR, Thomas NS, Orr S, Linch DC, Devereux S. 2000. Monocyte-derived dendritic cells do not proliferate and are not susceptible to retroviral transduction. Br J Haematol 108: 817–824. - PubMed
-
- Bruniquel D, Schwartz RH. 2003. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat Immunol 4: 235–240. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases