

The role of breast-feeding in infant immune system: a systems perspective on the intestinal microbiome
- PMID: 26399409
- PMCID: PMC4581423
- DOI: 10.1186/s40168-015-0104-7
The role of breast-feeding in infant immune system: a systems perspective on the intestinal microbiome
Abstract
Background: The human intestinal microbiota changes from being sparsely populated and variable to possessing a mature, adult-like stable microbiome during the first 2 years of life. This assembly process of the microbiota can lead to either negative or positive effects on health, depending on the colonization sequence and diet. An integrative study on the diet, the microbiota, and genomic activity at the transcriptomic level may give an insight into the role of diet in shaping the human/microbiome relationship. This study aims at better understanding the effects of microbial community and feeding mode (breast-fed and formula-fed) on the immune system, by comparing intestinal metagenomic and transcriptomic data from breast-fed and formula-fed babies.
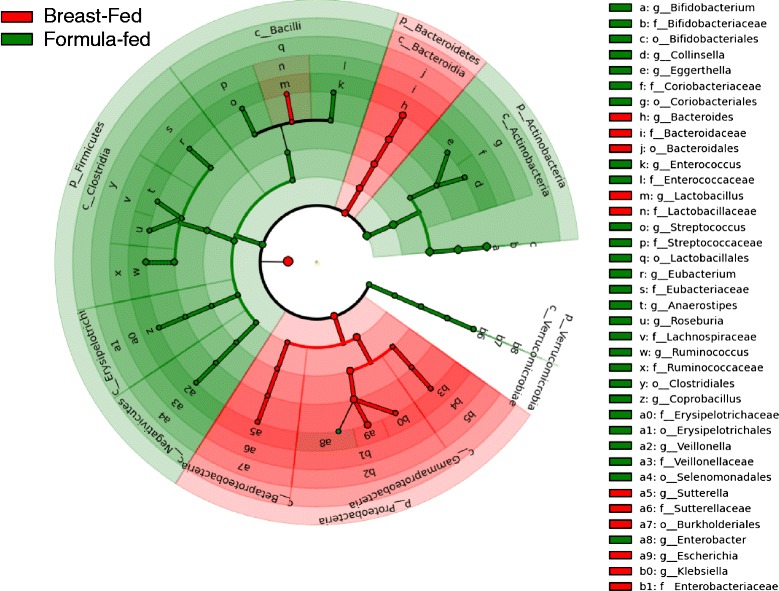
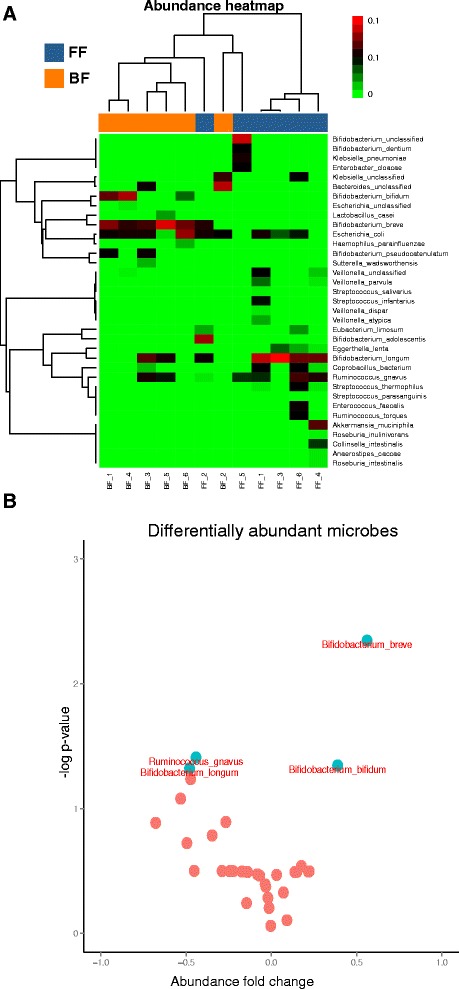
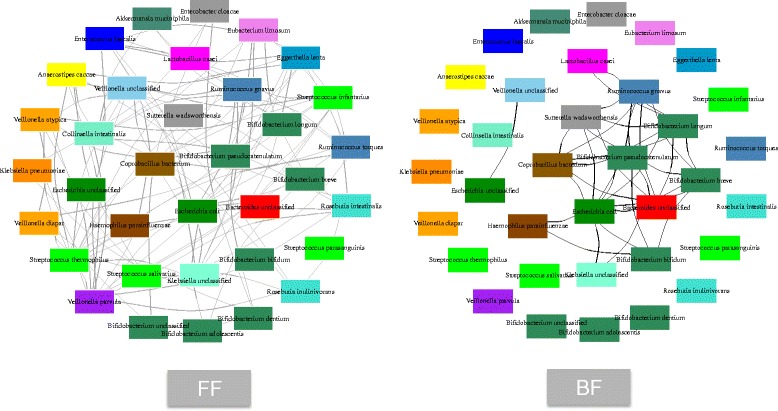
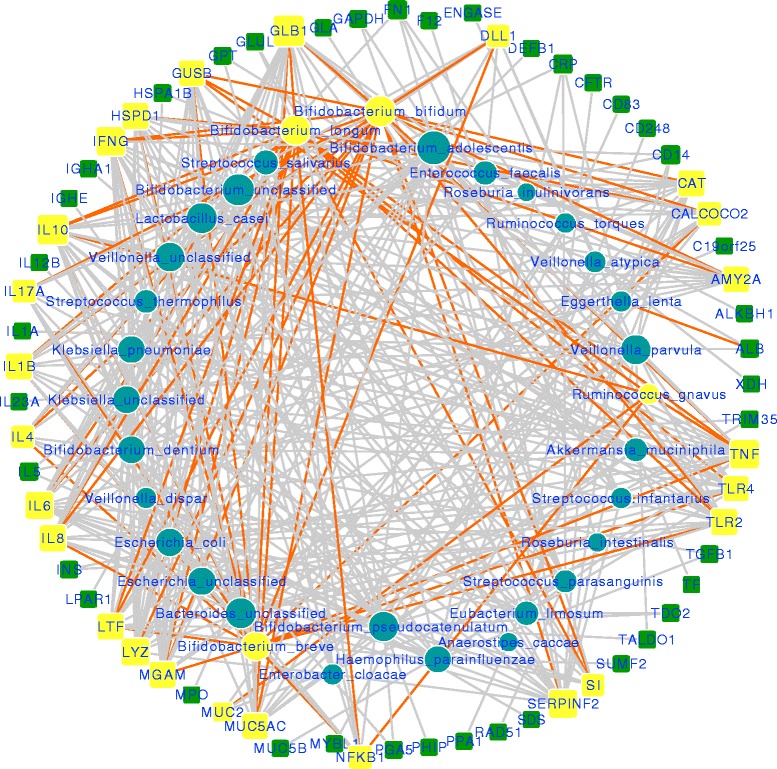
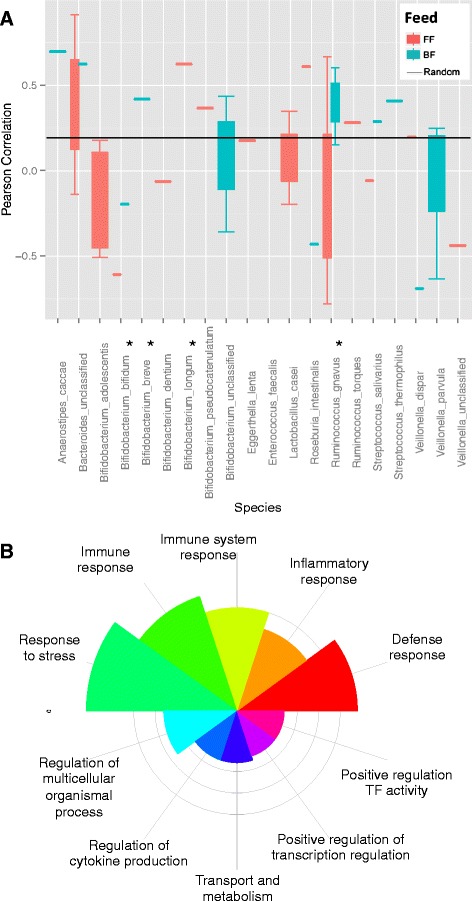
Results: We re-analyzed a published metagenomics and host gene expression dataset from a systems biology perspective. Our results show that breast-fed samples co-express genes associated with immunological, metabolic, and biosynthetic activities. The diversity of the microbiota is higher in formula-fed than breast-fed infants, potentially reflecting the weaker dependence of infants on maternal microbiome. We mapped the microbial composition and the expression patterns for host systems and studied their relationship from a systems biology perspective, focusing on the differences.
Conclusions: Our findings revealed that there is co-expression of more genes in breast-fed samples but lower microbial diversity compared to formula-fed. Applying network-based systems biology approach via enrichment of microbial species with host genes revealed the novel key relationships of the microbiota with immune and metabolic activity. This was supported statistically by data and literature.
Figures

References
-
- Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci. 2011;108(Suppl 1):4586–91. doi:10.1073/pnas.1000097107. http://www.pnas.org/content/108/Supplement_1/4586.long. - PMC - PubMed
-
- Claesson MJ, Jeffery IB, Conde S, Power SE, O’Connor EM, Cusack S, Harris HMB, Coakley M, Lakshminarayanan B, O’Sullivan O, Fitzgerald GF, Deane J, O’Connor M, Harnedy N, O’Connor K, O’Mahony D, van Sinderen D, Wallace M, Brennan L, Stanton C, Marchesi JR, Fitzgerald AP, Shanahan F, Hill C, Ross RP, O’Toole PW. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488(7410):178–84. doi: 10.1038/nature11319. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
