

Postsynaptic Depolarization Enhances GABA Drive to Dorsomedial Hypothalamic Neurons through Somatodendritic Cholecystokinin Release
- PMID: 26400945
- PMCID: PMC6605435
- DOI: 10.1523/JNEUROSCI.3123-14.2015
Postsynaptic Depolarization Enhances GABA Drive to Dorsomedial Hypothalamic Neurons through Somatodendritic Cholecystokinin Release
Abstract
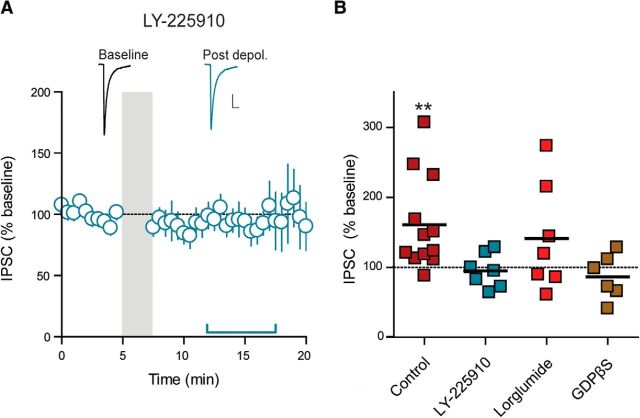
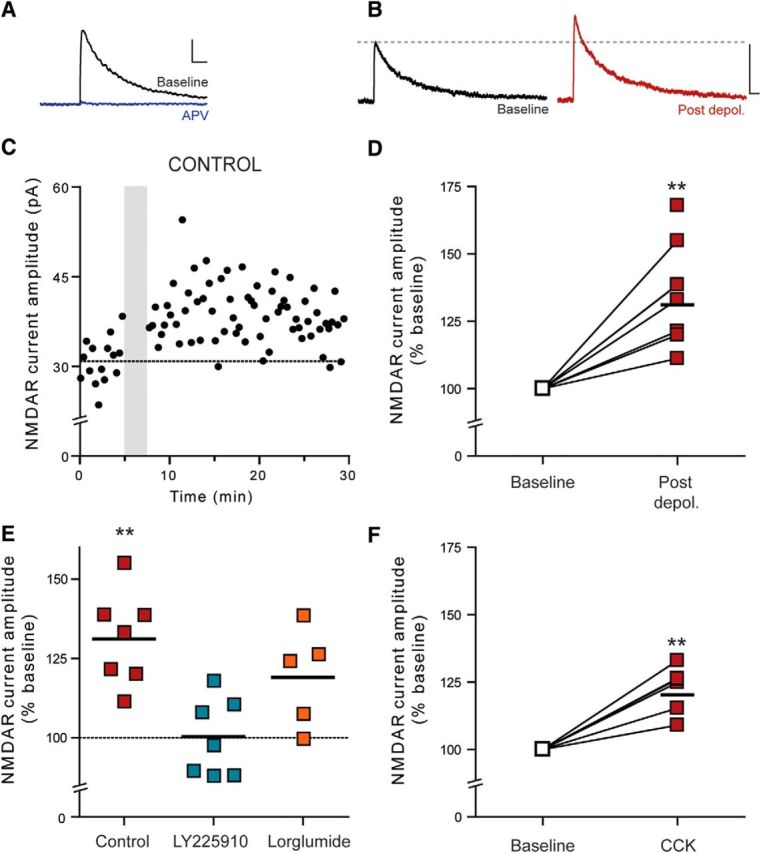
Somatodendritically released peptides alter synaptic function through a variety of mechanisms, including autocrine actions that liberate retrograde transmitters. Cholecystokinin (CCK) is a neuropeptide expressed in neurons in the dorsomedial hypothalamic nucleus (DMH), a region implicated in satiety and stress. There are clear demonstrations that exogenous CCK modulates food intake and neuropeptide expression in the DMH, but there is no information on how endogenous CCK alters synaptic properties. Here, we provide the first report of somatodendritic release of CCK in the brain in male Sprague Dawley rats. CCK is released from DMH neurons in response to repeated postsynaptic depolarizations, and acts in an autocrine fashion on CCK2 receptors to enhance postsynaptic NMDA receptor function and liberate the retrograde transmitter, nitric oxide (NO). NO subsequently acts presynaptically to enhance GABA release through a soluble guanylate cyclase-mediated pathway. These data provide the first demonstration of synaptic actions of somatodendritically released CCK in the hypothalamus and reveal a new form of retrograde plasticity, depolarization-induced potentiation of inhibition. Significance statement: Somatodendritic signaling using endocannabinoids or nitric oxide to alter the efficacy of afferent transmission is well established. Despite early convincing evidence for somatodendritic release of neurohypophysial peptides in the hypothalamus, there is only limited evidence for this mode of release for other peptides. Here, we provide the first evidence for somatodendritic release of the satiety peptide cholecystokinin (CCK) in the brain. We also reveal a new form of synaptic plasticity in which postsynaptic depolarization results in enhancement of inhibition through the somatodendritic release of CCK.
Keywords: GABA; NMDAR; cholecystokinin; dorsomedial hypothalamus; nitric oxide; somatodendritic release.
Copyright © 2015 the authors 0270-6474/15/3513160-11$15.00/0.
Figures





References
-
- Bi S, Ladenheim EE, Schwartz GJ, Moran TH. A role for NPY overexpression in the dorsomedial hypothalamus in hyperphagia and obesity of OLETF rats. Am J Physiol Regul Integr Comp Physiol. 2001;281:R254–R260. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources