

A novel mutation in TTC8 is associated with progressive retinal atrophy in the golden retriever
- PMID: 26401321
- PMCID: PMC4574394
- DOI: 10.1186/2052-6687-1-4
A novel mutation in TTC8 is associated with progressive retinal atrophy in the golden retriever
Abstract
Background: Generalized progressive retinal atrophy (PRA) is a group of inherited eye diseases characterised by progressive retinal degeneration that ultimately leads to blindness in dogs. To date, more than 20 different mutations causing canine-PRA have been described and several breeds including the Golden Retriever are affected by more than one form of PRA. Genetically distinct forms of PRA may have different clinical characteristics such as rate of progression and age of onset. However, in many instances the phenotype of different forms of PRA cannot be distinguished at the basic clinical level achieved during routine ophthalmoscopic examination. Mutations in two distinct genes have been reported to cause PRA in Golden Retrievers (prcd-PRA and GR_PRA1), but for approximately 39% of cases in this breed the causal mutation remains unknown.
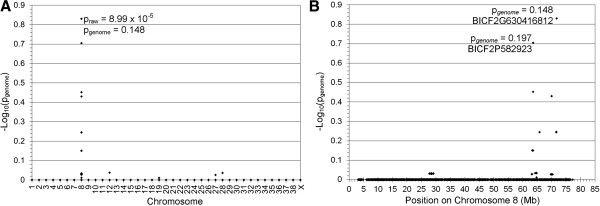
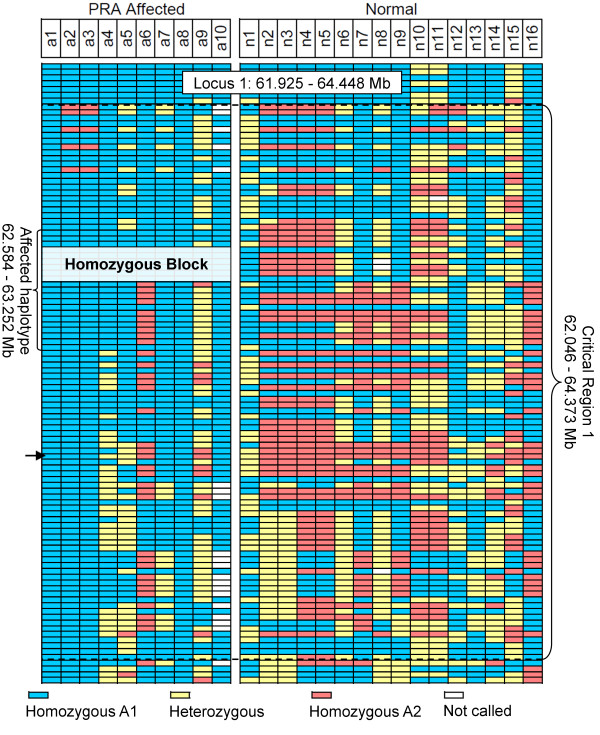
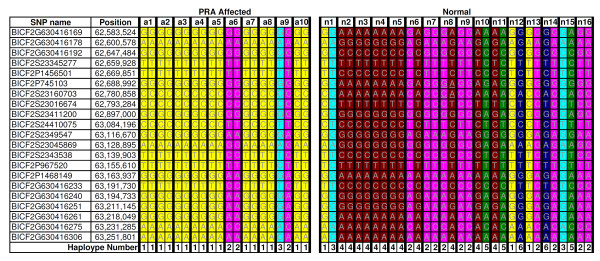
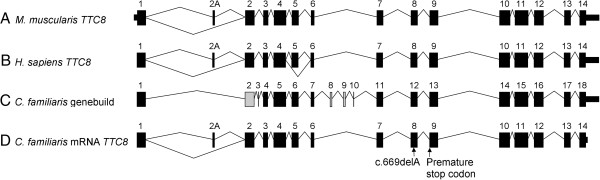
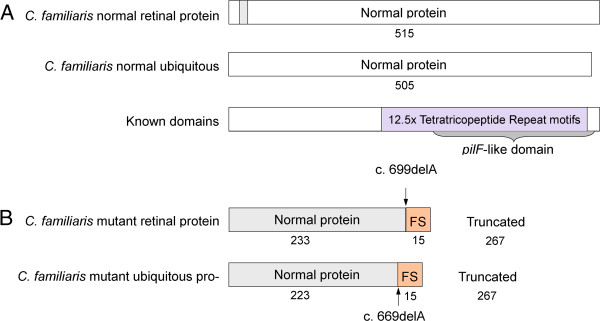
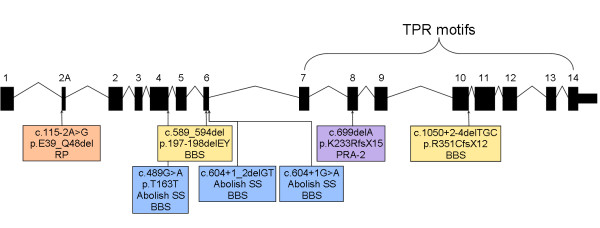
Results: A genome-wide association study of 10 PRA cases and 16 controls identified an association on chromosome 8 not previously associated with PRA (praw = 1.30×10(-6) and corrected with 100,000 permutations, pgenome = 0.148). Using haplotype analysis we defined a 737 kb critical region containing 6 genes. Two of the genes (TTC8 and SPATA7) have been associated with Retinitis Pigmentosa (RP) in humans. Using targeted next generation sequencing a single nucleotide deletion was identified in exon 8 of the TTC8 gene of affected Golden Retrievers. The frame shift mutation was predicted to cause a premature termination codon. In a larger cohort, this mutation, TTC8 c.669delA, segregates correctly in 22 out of 29 cases tested (75.9%). Of the PRA controls none are homozygous for the mutation, only 3.5% carry the mutation and 96.5% are homozygous wildtype.
Conclusions: Our results show that PRA is genetically heterogeneous in one of the world's numerically largest breeds, the Golden Retriever, and is caused by multiple, distinct mutations. Here we discuss the mutation that causes a form of PRA, that we have termed PRA2, that accounts for approximately 30% of PRA cases in the breed. The genetic explanation for approximately 9% of cases remains to be identified. PRA2 is a naturally occurring animal model for Retinitis Pigmentosa, and potentially Bardet-Biedl Syndrome.
Figures

References
-
- André C, Chaudieu G, Thomas A, Jongh O, Jegou JP, Chahory S, Clerc B, Pilorge P, Brenac O. Hereditary retinopathies in the dog: genetic fundamentals and genetic tests. Pratique Médicale et Chirurgicale de l’Animal de Compagnie. 2008;43:75–84. doi: 10.1016/j.anicom.2008.06.002. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
