

Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice
- PMID: 26404765
- PMCID: PMC4805450
- DOI: 10.1002/hep.28245
Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice
Abstract
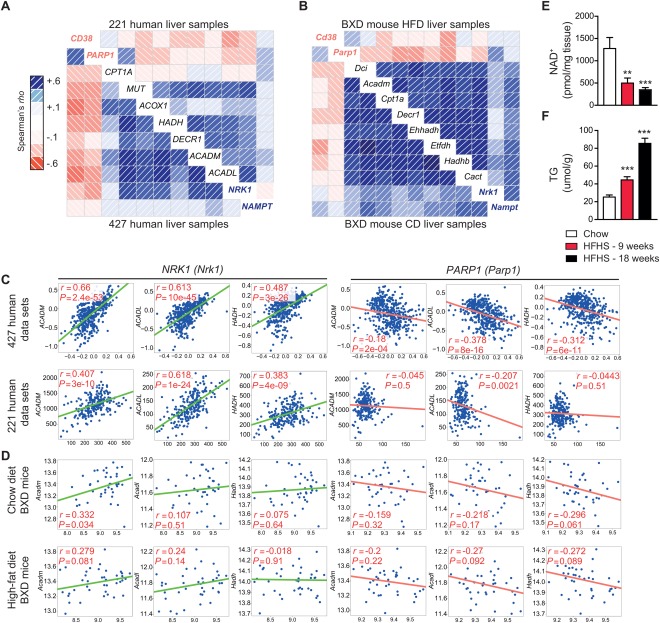
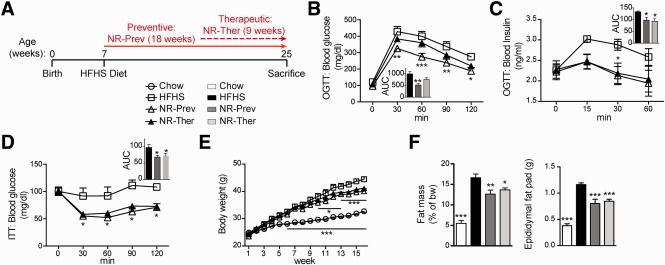
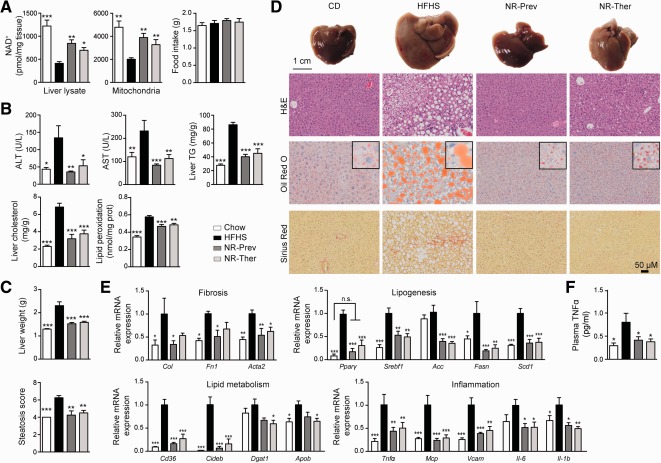
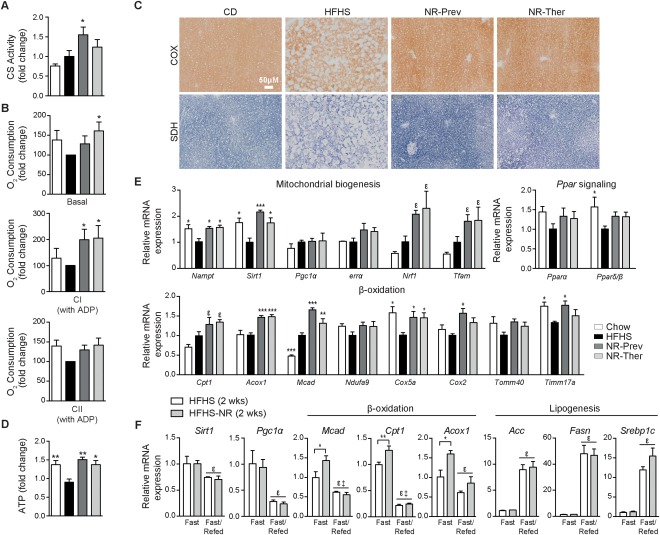
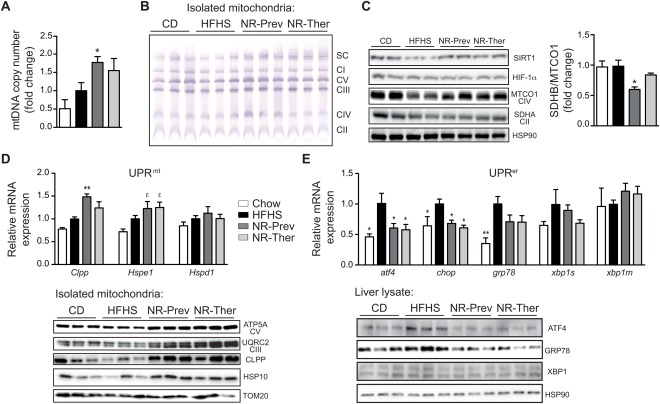
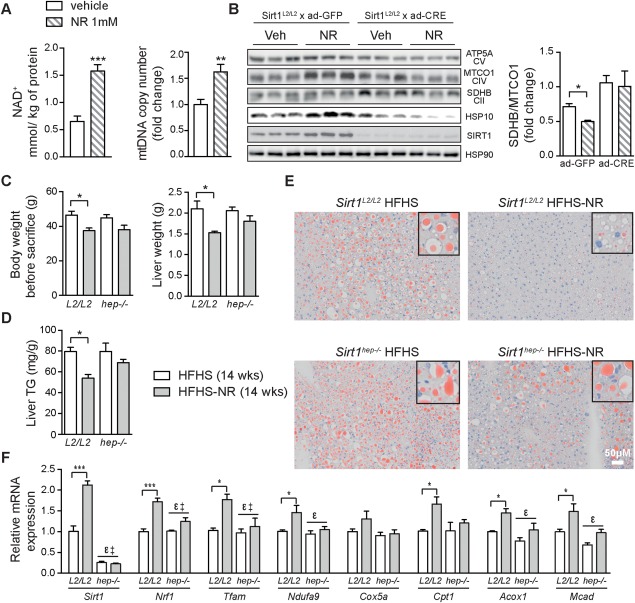
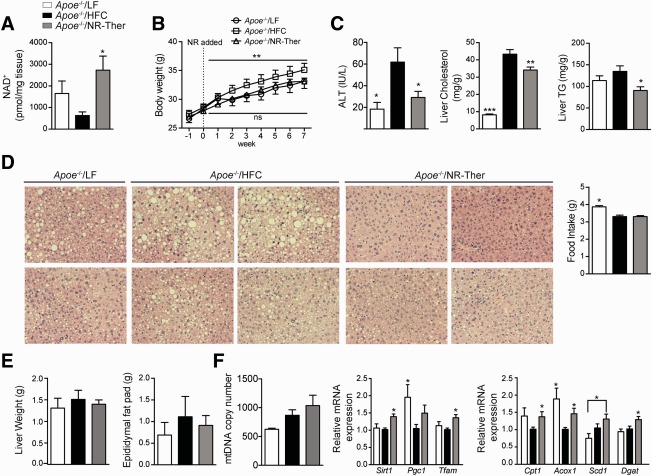
With no approved pharmacological treatment, nonalcoholic fatty liver disease (NAFLD) is now the most common cause of chronic liver disease in Western countries and its worldwide prevalence continues to increase along with the growing obesity epidemic. Here, we show that a high-fat high-sucrose (HFHS) diet, eliciting chronic hepatosteatosis resembling human fatty liver, lowers hepatic nicotinamide adenine dinucleotide (NAD(+) ) levels driving reductions in hepatic mitochondrial content, function, and adenosine triphosphate (ATP) levels, in conjunction with robust increases in hepatic weight, lipid content, and peroxidation in C57BL/6J mice. To assess the effect of NAD(+) repletion on the development of steatosis in mice, nicotinamide riboside, a precursor of NAD(+) biosynthesis, was added to the HFHS diet, either as a preventive strategy or as a therapeutic intervention. We demonstrate that NR prevents and reverts NAFLD by inducing a sirtuin (SIRT)1- and SIRT3-dependent mitochondrial unfolded protein response, triggering an adaptive mitohormetic pathway to increase hepatic β-oxidation and mitochondrial complex content and activity. The cell-autonomous beneficial component of NR treatment was revealed in liver-specific Sirt1 knockout mice (Sirt1(hep-/-) ), whereas apolipoprotein E-deficient mice (Apoe(-/-) ) challenged with a high-fat high-cholesterol diet affirmed the use of NR in other independent models of NAFLD.
Conclusion: Our data warrant the future evaluation of NAD(+) boosting strategies to manage the development or progression of NAFLD.
© 2015 The Authors. Hepatology published by Wiley Periodicals, Inc., on behalf of the American Association for the Study of Liver Diseases.
Figures
Comment in
-
Therapeutic potential of nicotinamide adenine dinucleotide for nonalcoholic fatty liver disease.Hepatology. 2016 Apr;63(4):1074-7. doi: 10.1002/hep.28383. Epub 2016 Feb 16. Hepatology. 2016. PMID: 26661503 No abstract available.
References
-
- Musso G, Cassader M, Rosina F, Gambino R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non‐alcoholic fatty liver disease (NAFLD): a systematic review and meta‐analysis of randomised trials. Diabetologia 2012;55:885‐904. - PubMed
-
- Sanyal AJ, Campbell‐Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001;120:1183‐1192. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
