

Remote control of therapeutic T cells through a small molecule-gated chimeric receptor
- PMID: 26405231
- PMCID: PMC4721629
- DOI: 10.1126/science.aab4077
Remote control of therapeutic T cells through a small molecule-gated chimeric receptor
Abstract
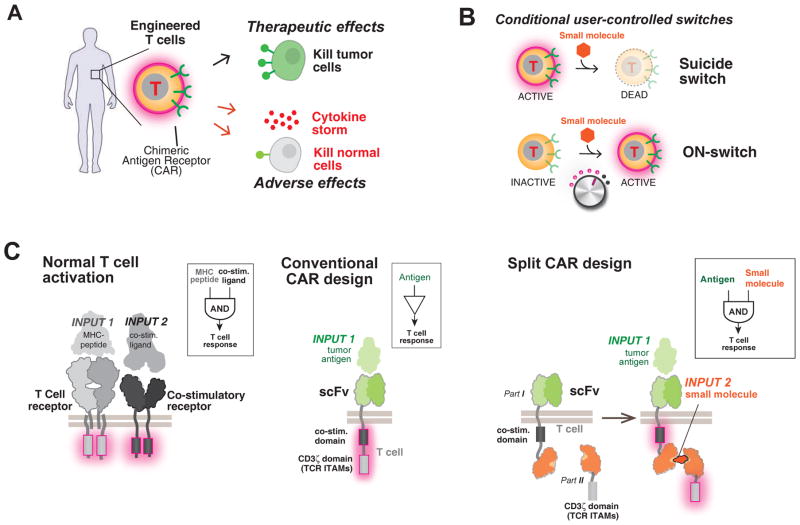
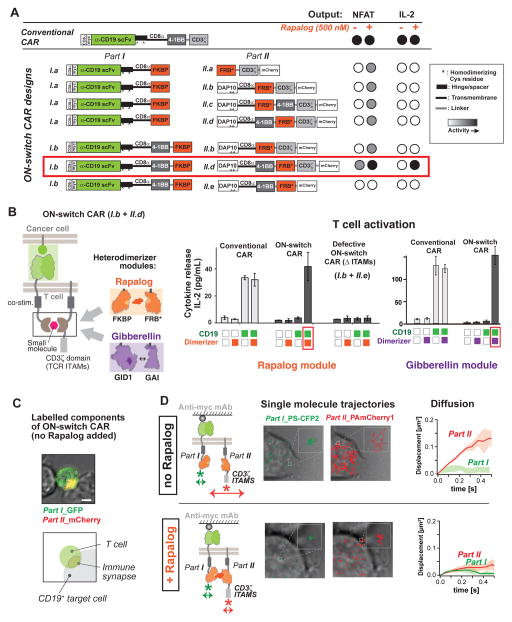
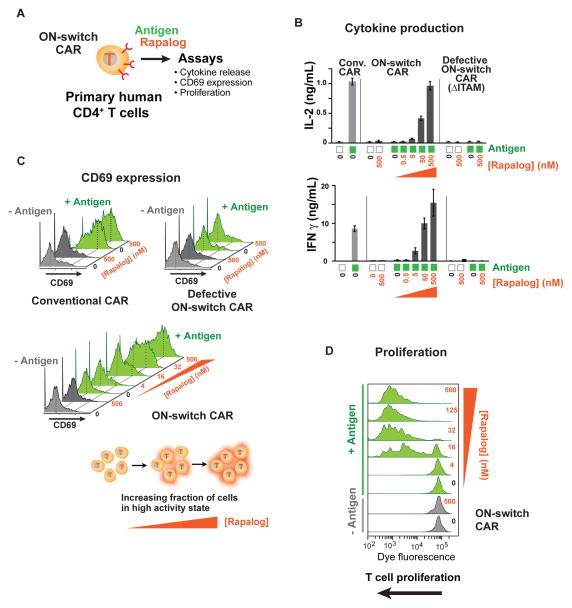
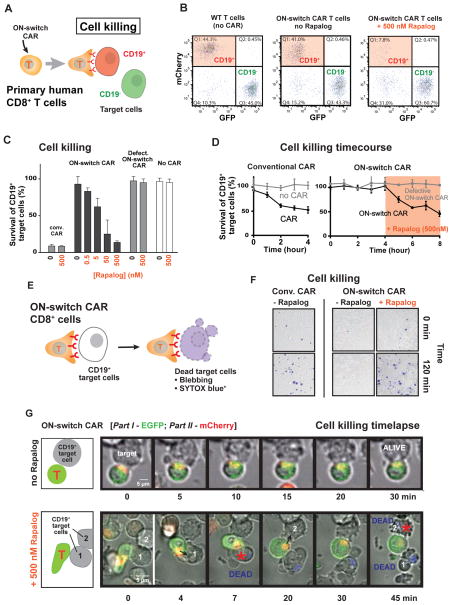
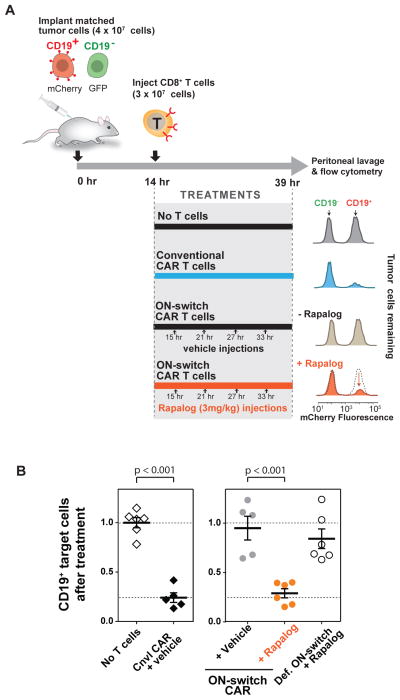
There is growing interest in using engineered cells as therapeutic agents. For example, synthetic chimeric antigen receptors (CARs) can redirect T cells to recognize and eliminate tumor cells expressing specific antigens. Despite promising clinical results, these engineered T cells can exhibit excessive activity that is difficult to control and can cause severe toxicity. We designed "ON-switch" CARs that enable small-molecule control over T cell therapeutic functions while still retaining antigen specificity. In these split receptors, antigen-binding and intracellular signaling components assemble only in the presence of a heterodimerizing small molecule. This titratable pharmacologic regulation could allow physicians to precisely control the timing, location, and dosage of T cell activity, thereby mitigating toxicity. This work illustrates the potential of combining cellular engineering with orthogonal chemical tools to yield safer therapeutic cells that tightly integrate cell-autonomous recognition and user control.
Copyright © 2015, American Association for the Advancement of Science.
Figures
Comment in
-
Immunotherapy: Remote control CARs.Nat Rev Immunol. 2015 Dec;15(12):726-7. doi: 10.1038/nri3938. Epub 2015 Nov 6. Nat Rev Immunol. 2015. PMID: 26542635 No abstract available.
-
The quest for spatio-temporal control of CAR T cells.Cell Res. 2015 Dec;25(12):1281-2. doi: 10.1038/cr.2015.131. Epub 2015 Nov 17. Cell Res. 2015. PMID: 26575974 Free PMC article.
-
Immunotherapy: Remote control CARs.Nat Rev Drug Discov. 2015 Dec;14(12):819. doi: 10.1038/nrd4789. Nat Rev Drug Discov. 2015. PMID: 26620410 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
