

Evaluating the Performance of Fine-Mapping Strategies at Common Variant GWAS Loci
- PMID: 26406328
- PMCID: PMC4583479
- DOI: 10.1371/journal.pgen.1005535
Evaluating the Performance of Fine-Mapping Strategies at Common Variant GWAS Loci
Abstract
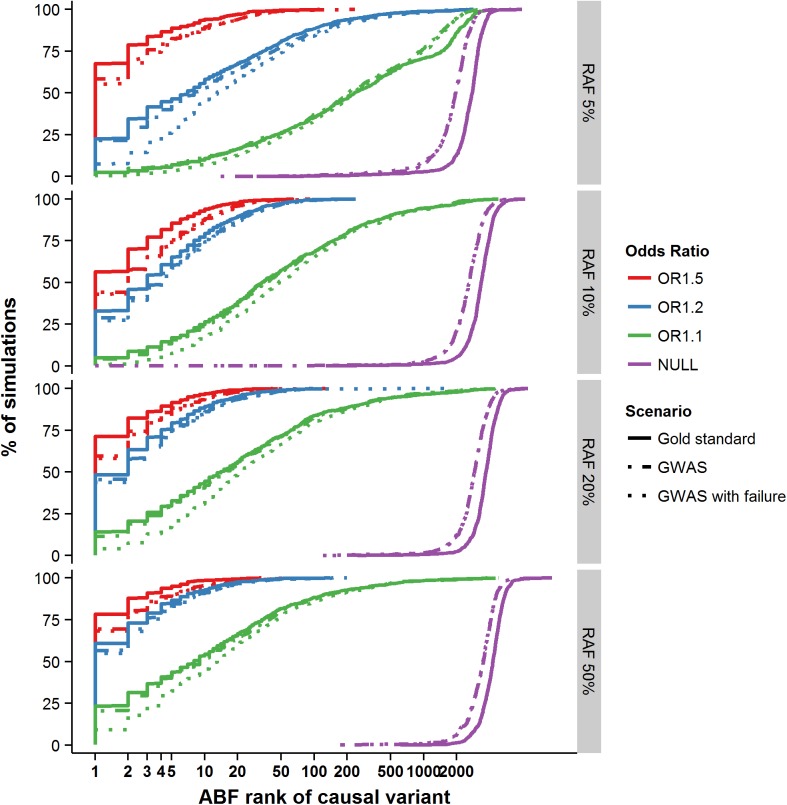
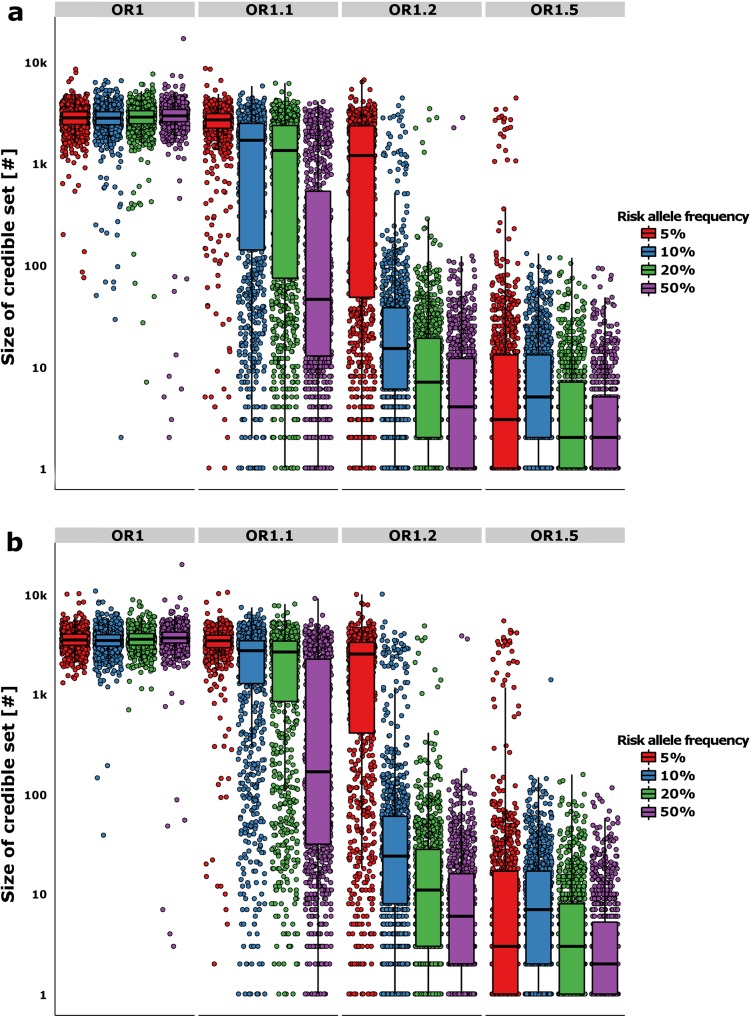
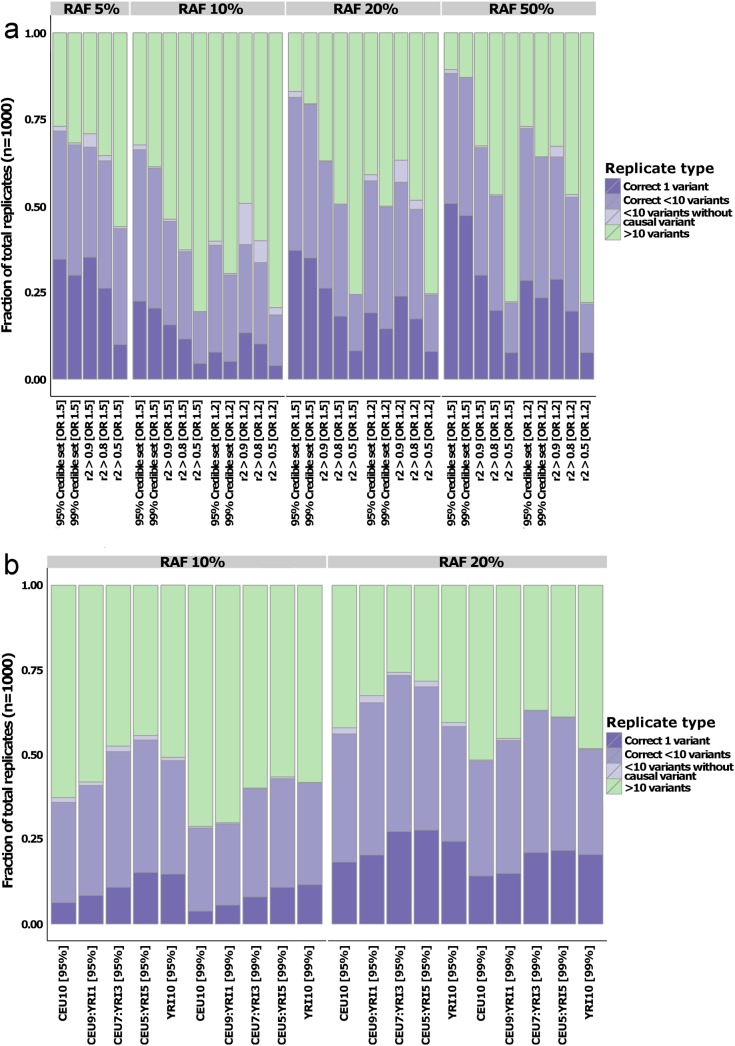
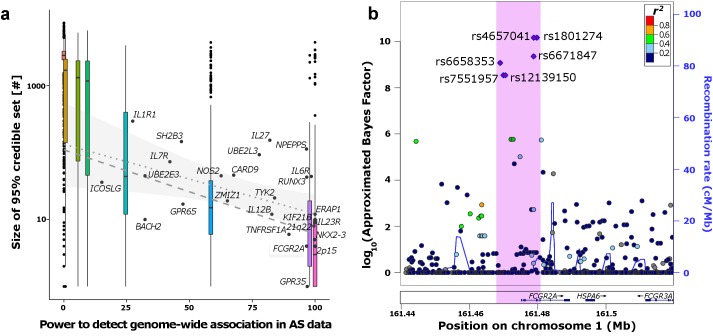
The growing availability of high-quality genomic annotation has increased the potential for mechanistic insights when the specific variants driving common genome-wide association signals are accurately localized. A range of fine-mapping strategies have been advocated, and specific successes reported, but the overall performance of such approaches, in the face of the extensive linkage disequilibrium that characterizes the human genome, is not well understood. Using simulations based on sequence data from the 1000 Genomes Project, we quantify the extent to which fine-mapping, here conducted using an approximate Bayesian approach, can be expected to lead to useful improvements in causal variant localization. We show that resolution is highly variable between loci, and that performance is severely degraded as the statistical power to detect association is reduced. We confirm that, where causal variants are shared between ancestry groups, further improvements in performance can be obtained in a trans-ethnic fine-mapping design. Finally, using empirical data from a recently published genome-wide association study for ankylosing spondylitis, we provide empirical confirmation of the behaviour of the approximate Bayesian approach and demonstrate that seven of twenty-six loci can be fine-mapped to fewer than ten variants.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
