

DYRK2 Negatively Regulates Type I Interferon Induction by Promoting TBK1 Degradation via Ser527 Phosphorylation
- PMID: 26407194
- PMCID: PMC4583546
- DOI: 10.1371/journal.ppat.1005179
DYRK2 Negatively Regulates Type I Interferon Induction by Promoting TBK1 Degradation via Ser527 Phosphorylation
Abstract
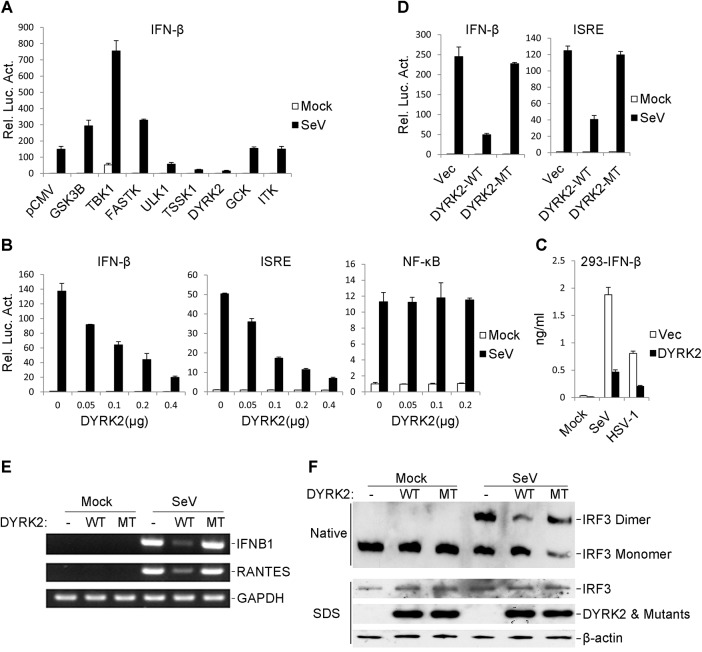
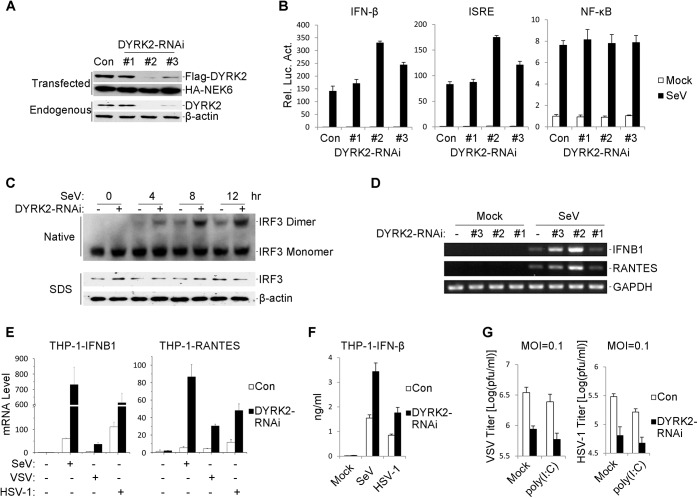
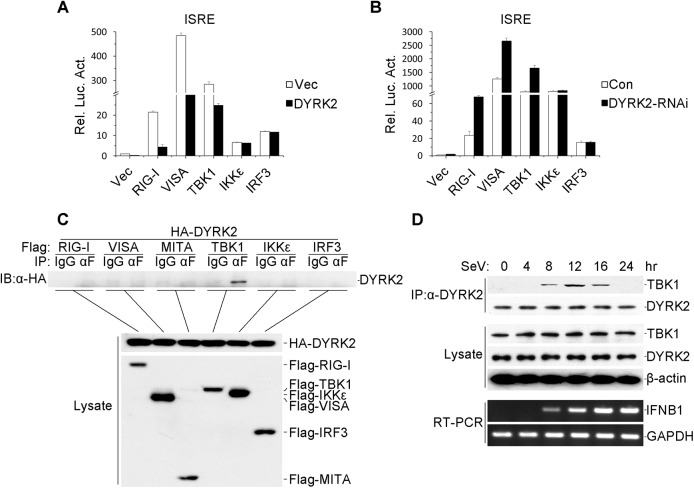
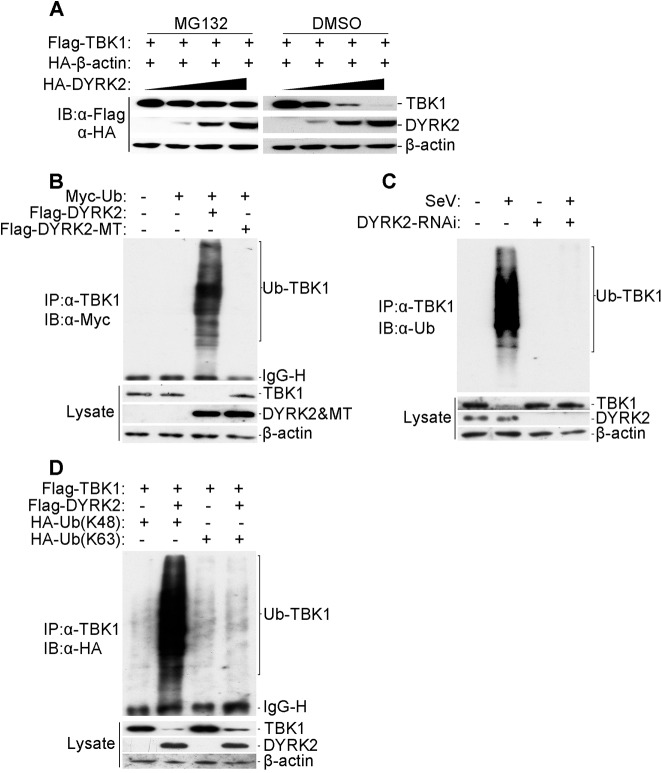
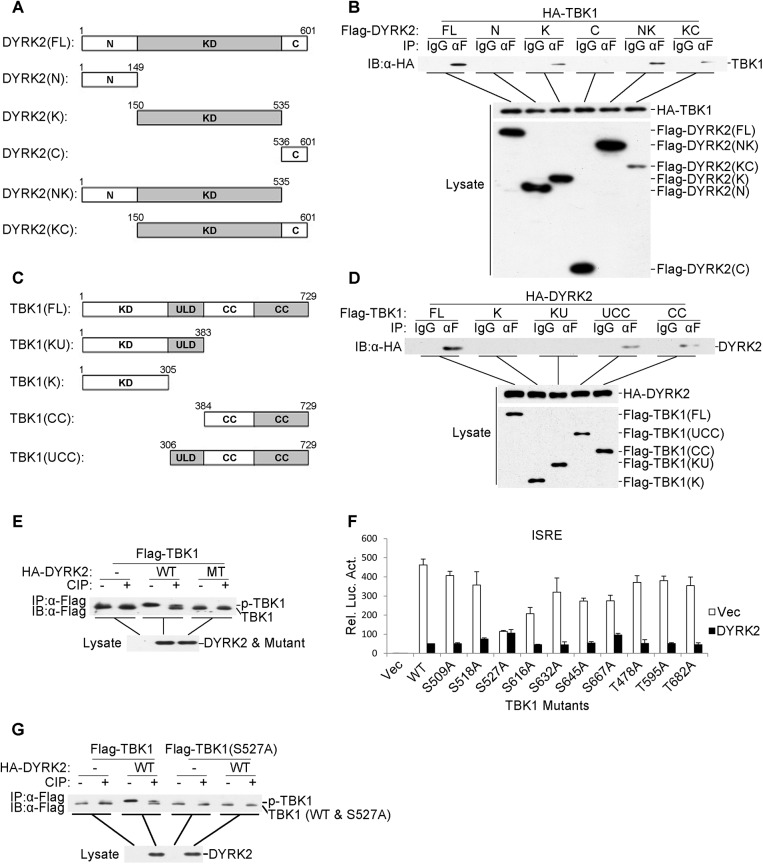
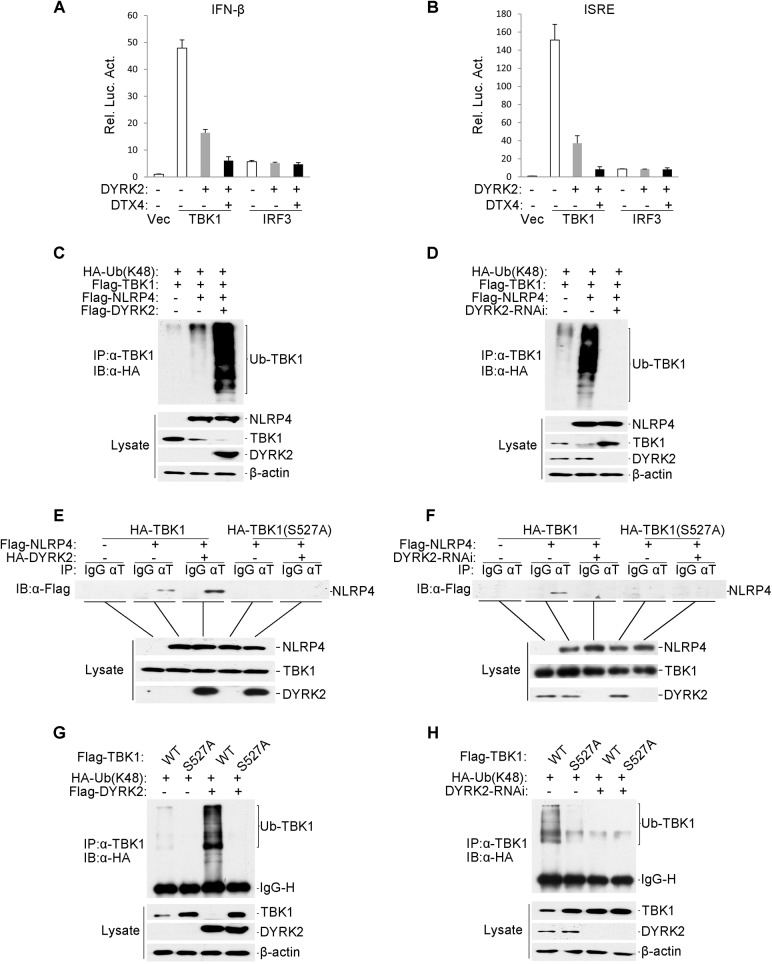
Viral infection activates the transcription factors NF-κB and IRF3, which contribute to the induction of type I interferons (IFNs) and cellular antiviral responses. Protein kinases play a critical role in various signaling pathways by phosphorylating their substrates. Here, we identified dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 2 (DYRK2) as a negative regulator of virus-triggered type I IFN induction. DYRK2 inhibited the virus-triggered induction of type I IFNs and promoted the K48-linked ubiquitination and degradation of TANK-binding kinase 1 (TBK1) in a kinase-activity-dependent manner. We further found that DYRK2 phosphorylated Ser527 of TBK1, which is essential for the recruitment of NLRP4 and for the E3 ubiquitin ligase DTX4 to degrade TBK1. These findings suggest that DYRK2 negatively regulates virus-triggered signaling by targeting TBK1 for phosphorylation and priming it for degradation, and these data provide new insights into the molecular mechanisms that dictate the cellular antiviral response.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
