

Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape
- PMID: 26408185
- PMCID: PMC4582738
- DOI: 10.1186/s13059-015-0769-z
Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape
Erratum in
-
Erratum to: Deep sequencing and de novo assembly of the mouse occyte transcriptome define the contribution of transcription to the DNA methylation landscape.Genome Biol. 2015 Dec 3;16:271. doi: 10.1186/s13059-015-0809-8. Genome Biol. 2015. PMID: 26635312 Free PMC article. No abstract available.
Abstract
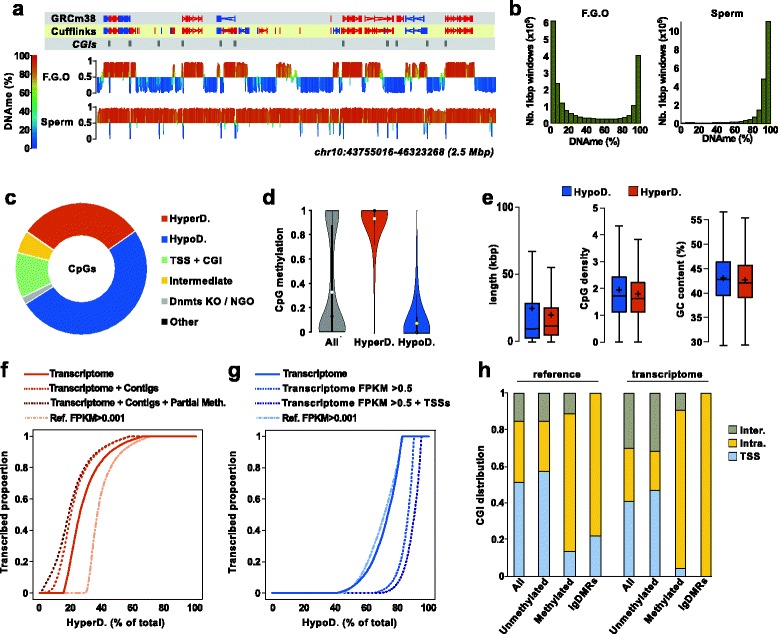
Background: Previously, a role was demonstrated for transcription in the acquisition of DNA methylation at imprinted control regions in oocytes. Definition of the oocyte DNA methylome by whole genome approaches revealed that the majority of methylated CpG islands are intragenic and gene bodies are hypermethylated. Yet, the mechanisms by which transcription regulates DNA methylation in oocytes remain unclear. Here, we systematically test the link between transcription and the methylome.
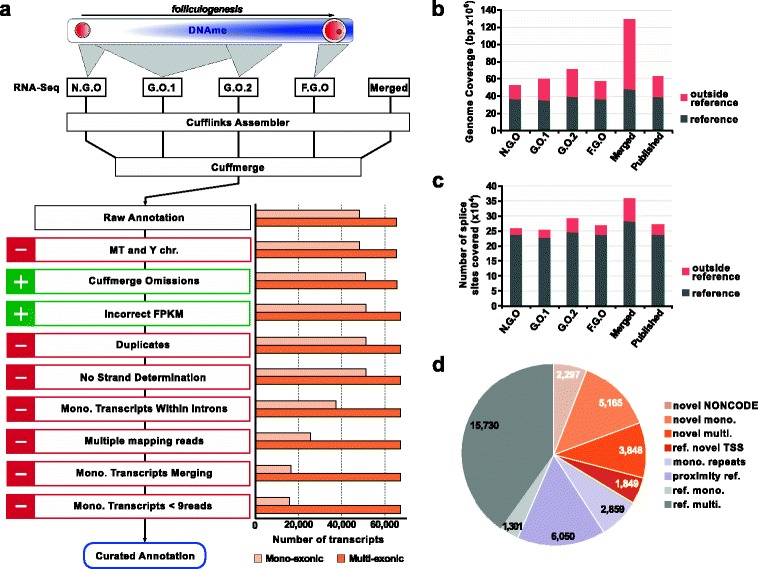
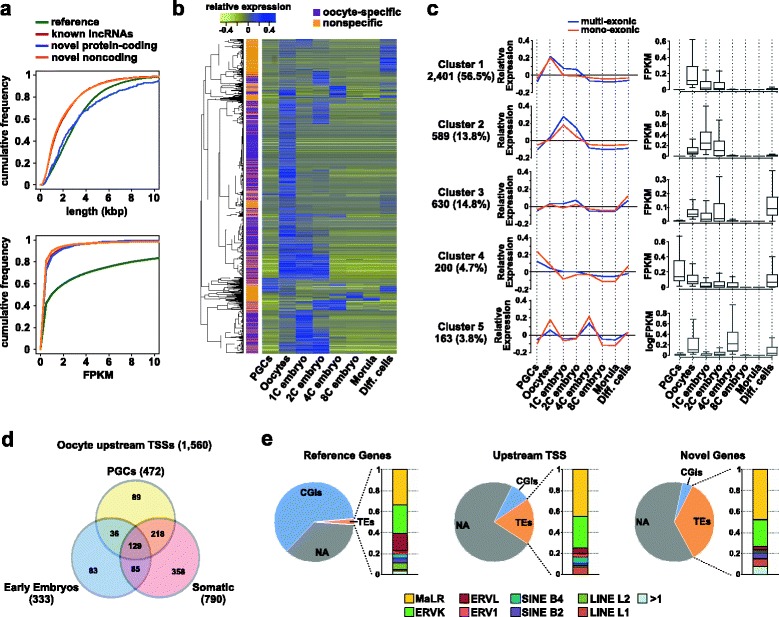
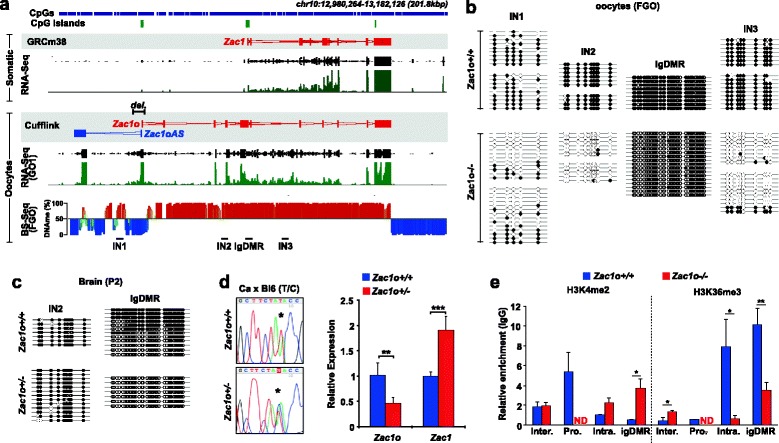
Results: We perform deep RNA-Seq and de novo transcriptome assembly at different stages of mouse oogenesis. This reveals thousands of novel non-annotated genes, as well as alternative promoters, for approximately 10 % of reference genes expressed in oocytes. In addition, a large fraction of novel promoters coincide with MaLR and ERVK transposable elements. Integration with our transcriptome assembly reveals that transcription correlates accurately with DNA methylation and accounts for approximately 85-90 % of the methylome. We generate a mouse model in which transcription across the Zac1/Plagl1 locus is abrogated in oocytes, resulting in failure of DNA methylation establishment at all CpGs of this locus. ChIP analysis in oocytes reveals H3K4me2 enrichment at the Zac1 imprinted control region when transcription is ablated, establishing a connection between transcription and chromatin remodeling at CpG islands by histone demethylases.
Conclusions: By precisely defining the mouse oocyte transcriptome, this work not only highlights transcription as a cornerstone of DNA methylation establishment in female germ cells, but also provides an important resource for developmental biology research.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
