

Hyperprolinemia in Type 2 Glutaric Aciduria and MADD-Like Profiles
- PMID: 26409463
- PMCID: PMC4864717
- DOI: 10.1007/8904_2015_481
Hyperprolinemia in Type 2 Glutaric Aciduria and MADD-Like Profiles
Abstract
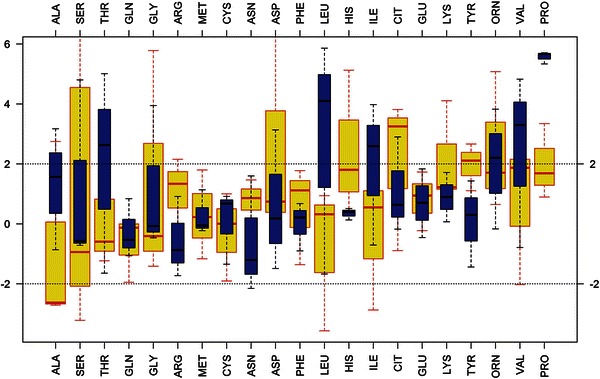
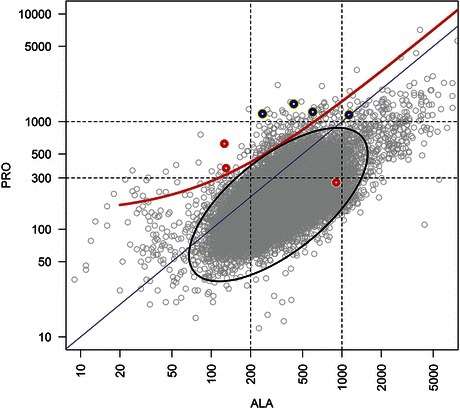
Classical neonatal-onset glutaric aciduria type 2 (MAD deficiency) is a severe disorder of mitochondrial fatty acid oxidation associated with poor survival. Secondary dysfunction of acyl-CoA dehydrogenases may result from deficiency for riboflavin transporters, leading to severe disorders that, nevertheless, are treatable by riboflavin supplementation. In the last 10 years, we identified nine newborns with biochemical features consistent with MAD deficiency, only four of whom survived past the neonatal period. A likely iatrogenic cause of riboflavin deficiency was found in two premature newborns having parenteral nutrition, one of whom recovered upon multivitamin supplementation, whereas the other died before diagnosis. Four other patients had demonstrated mutations involving ETF or ETF-DH flavoproteins, whereas the remaining three patients presumably had secondary deficiencies of unknown mechanism. Interestingly, six newborns among the seven tested for plasma amino acids had pronounced hyperprolinemia. In one case, because the initial diagnostic workup did not include organic acids and acylcarnitine profiling, clinical presentation and hyperprolinemia suggested the diagnosis. Analysis of our full cohort of >50,000 samples from >30,000 patients suggests that the proline/alanine ratio may be a good marker of MAD deficiency and could contribute to a more effective management of the treatable forms.
Figures
References
-
- Bosch AM, Abeling NGGM, Ijlst L, et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis. 2011;34:159–164. doi: 10.1007/s10545-010-9242-z. - DOI - PMC - PubMed
-
- Frerman FE, Goodman SI, et al. Defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: glutaric acidemia type II. In: Scriver CR, Sly WS, Childs B, et al., editors. The metabolic and molecular basis of inherited disease. New York: McGraw-Hill; 2001. pp. 2357–2365.
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
