

Functional Insights into Chromatin Remodelling from Studies on CHARGE Syndrome
- PMID: 26411921
- PMCID: PMC4604214
- DOI: 10.1016/j.tig.2015.05.009
Functional Insights into Chromatin Remodelling from Studies on CHARGE Syndrome
Abstract
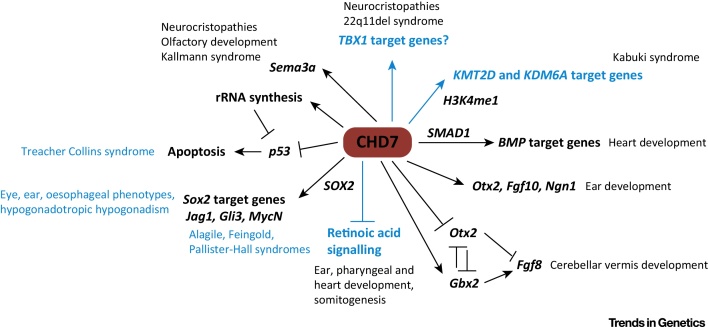
CHARGE syndrome is a rare genetic syndrome characterised by a unique combination of multiple organ anomalies. Dominant loss-of-function mutations in the gene encoding chromodomain helicase DNA binding protein 7 (CHD7), which is an ATP-dependent chromatin remodeller, have been identified as the cause of CHARGE syndrome. Here, we review recent work aimed at understanding the mechanism of CHD7 function in normal and pathological states, highlighting results from biochemical and in vivo studies. The emerging picture from this work suggests that the mechanisms by which CHD7 fine-tunes gene expression are context specific, consistent with the pleiotropic nature of CHARGE syndrome.
Keywords: CHARGE syndrome; CHD7; chromatin remodelling; congenital disease; epigenetic mechanisms.
Copyright © 2015 Elsevier Ltd. All rights reserved.
Figures
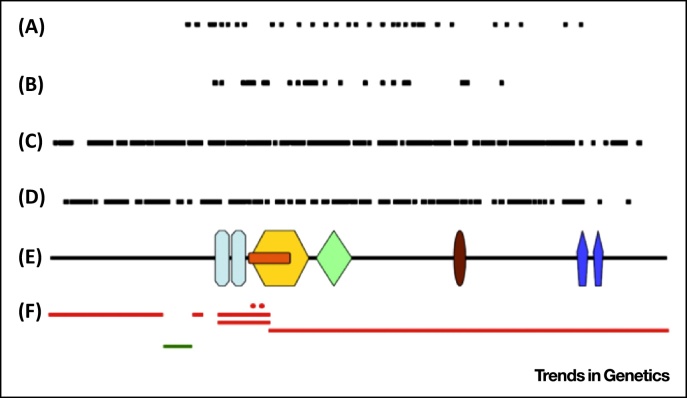
 chromodomain;
chromodomain;  helicase N;
helicase N;  DEXDc;
DEXDc;  Helicase C;
Helicase C;  SANT domain;
SANT domain;  BRK domain. Adapted from . An overview of mutations and polymorphic variants of the CHD7 gene can be found in the CHD7 locus-specific databasei.
BRK domain. Adapted from . An overview of mutations and polymorphic variants of the CHD7 gene can be found in the CHD7 locus-specific databasei.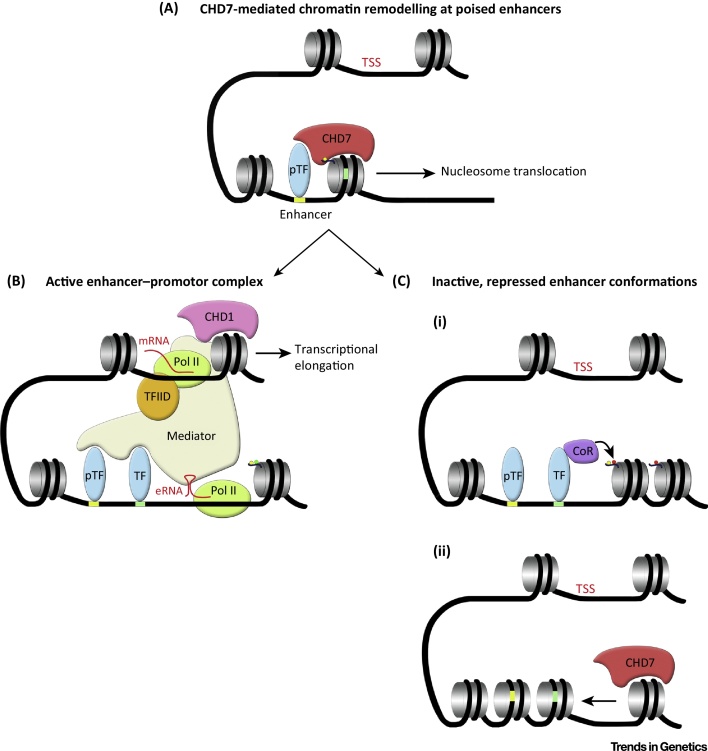
References
-
- Boycott K.M. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat. Rev. Genet. 2013;14:681–691. - PubMed
-
- Vissers L.E. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004;36:955–957. - PubMed
-
- Lalani S.R. CHARGE syndrome. In: Pagon R.A., editor. GeneReviews. University of Washington; 2012.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
