

Genetics of Progressive Supranuclear Palsy
- PMID: 26413239
- PMCID: PMC4572662
- DOI: 10.14802/jmd.15033
Genetics of Progressive Supranuclear Palsy
Abstract
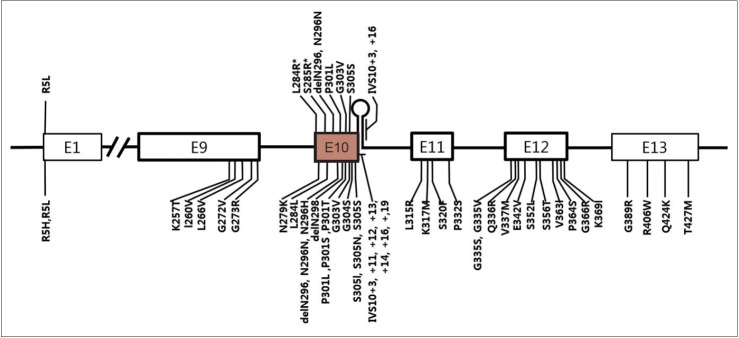
Progressive supranuclear palsy (PSP) is a neurodegenerative syndrome that is clinically characterized by progressive postural instability, supranuclear gaze palsy, parkinsonism and cognitive decline. Pathologically, diagnosis of PSP is based on characteristic features, such as neurofibrillary tangles, neutrophil threads, tau-positive astrocytes and their processes in basal ganglia and brainstem, and the accumulation of 4 repeat tau protein. PSP is generally recognized as a sporadic disorder; however, understanding of genetic background of PSP has been expanding rapidly. Here we review relevant publications to outline the genetics of PSP. Although only small number of familial PSP cases have been reported, the recognition of familial PSP has been increasing. In some familial cases of clinically probable PSP, PSP pathologies were confirmed based on NINDS neuropathological diagnostic criteria. Several mutations in MAPT, the gene that causes a form of familial frontotemporal lobar degeneration with tauopathy, have been identified in both sporadic and familial PSP cases. The H1 haplotype of MAPT is a risk haplotype for PSP, and within H1, a sub-haplotype (H1c) is associated with PSP. A recent genome-wide association study on autopsyproven PSP revealed additional PSP risk alleles in STX6 and EIF2AK3. Several heredodegenerative parkinsonian disorders are referred to as PSP-look-alikes because their clinical phenotype, but not their pathology, mimics PSP. Due to the fast development of genomics and bioinformatics, more genetic factors related to PSP are expected to be discovered. Undoubtedly, these studies will provide a better understanding of the pathogenesis of PSP and clues for developing therapeutic strategies.
Keywords: Familial progressive supranuclear palsy; Genetics; MAPT; Microtubule-associated protein tau; Progressive supranuclear palsy.
Conflict of interest statement
The authors have no financial conflicts of interest.
Figures
References
-
- Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol. 1964;10:333–359. - PubMed
-
- Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47:1–9. - PubMed
-
- Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy) Neurology. 1994;44:2015–2019. - PubMed
-
- Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol. 2010;23:394–400. - PubMed
-
- Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain. 2005;128(Pt 6):1247–1258. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
