

Altered Neuroinflammation and Behavior after Traumatic Brain Injury in a Mouse Model of Alzheimer's Disease
- PMID: 26414955
- PMCID: PMC4971425
- DOI: 10.1089/neu.2015.3970
Altered Neuroinflammation and Behavior after Traumatic Brain Injury in a Mouse Model of Alzheimer's Disease
Abstract
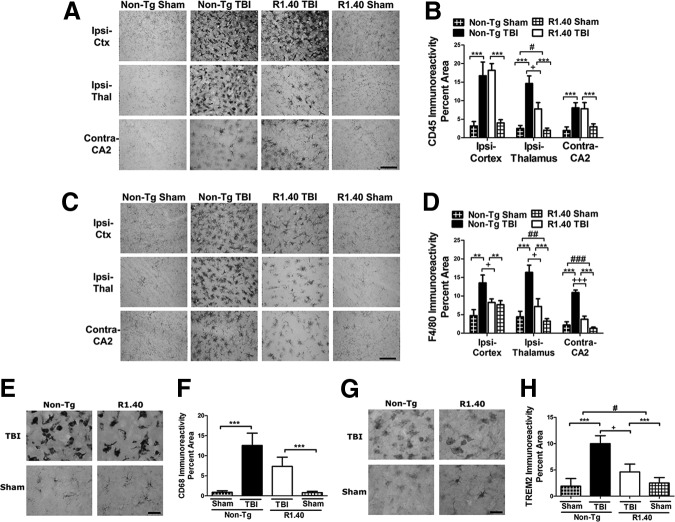
Traumatic brain injury (TBI) has acute and chronic sequelae, including an increased risk for the development of Alzheimer's disease (AD). TBI-associated neuroinflammation is characterized by activation of brain-resident microglia and infiltration of monocytes; however, recent studies have implicated beta-amyloid as a major manipulator of the inflammatory response. To examine neuroinflammation after TBI and development of AD-like features, these studies examined the effects of TBI in the presence and absence of beta-amyloid. The R1.40 mouse model of cerebral amyloidosis was used, with a focus on time points well before robust AD pathologies. Unexpectedly, in R1.40 mice, the acute neuroinflammatory response to TBI was strikingly muted, with reduced numbers of CNS myeloid cells acquiring a macrophage phenotype and decreased expression of inflammatory cytokines. At chronic time points, macrophage activation substantially declined in non-Tg TBI mice; however, it was relatively unchanged in R1.40 TBI mice. The persistent inflammatory response coincided with significant tissue loss between 3 and 120 days post-injury in R1.40 TBI mice, which was not observed in non-Tg TBI mice. Surprisingly, inflammatory cytokine expression was enhanced in R1.40 mice compared with non-Tg mice, regardless of injury group. Although R1.40 TBI mice demonstrated task-specific deficits in cognition, overall functional recovery was similar to non-Tg TBI mice. These findings suggest that accumulating beta-amyloid leads to an altered post-injury macrophage response at acute and chronic time points. Together, these studies emphasize the role of post-injury neuroinflammation in regulating long-term sequelae after TBI and also support recent studies implicating beta-amyloid as an immunomodulator.
Keywords: Alzheimer's disease; macrophage; neuroinflammation; traumatic brain injury.
Figures






References
-
- Nemetz P.N., Leibson C., Naessens J.M., Beard M., Kokmen E., Annegers J.F., and Kurland L.T. (1999). Traumatic brain injury and time to onset of Alzheimer's disease: a population-based study. Am. J. Epidemiol. 149, 32–40 - PubMed
-
- Plassman B.L., Havlik R.J., Steffens D.C., Helms M.J., Newman T.N., Drosdick D., Phillips C., Gau B.A., Welsh-Bohmer K.A., Burke J.R., Guralnik J.M., and Breitner J.C. (2000). Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 55, 1158–1166 - PubMed
-
- Mortimer J.A., van Duijn C.M., Chandra V., Fratiglioni L., Graves A.B., Heyman A., Jorm A.F., Kokmen E., Kondo K., Rocca W.A., Shalat S.L., Soininen H., and Hofman A. (1991). Head trauma as a risk factor for Alzheimer's disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int. J. Epidemiol. 20, Suppl 2, S28–S35 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
