

A genome wide transcriptional model of the complex response to pre-TCR signalling during thymocyte differentiation
- PMID: 26415229
- PMCID: PMC4745683
- DOI: 10.18632/oncotarget.5796
A genome wide transcriptional model of the complex response to pre-TCR signalling during thymocyte differentiation
Abstract
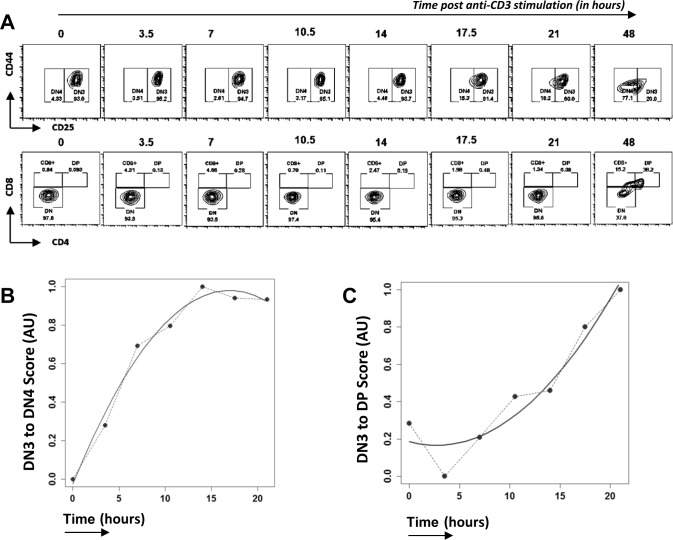
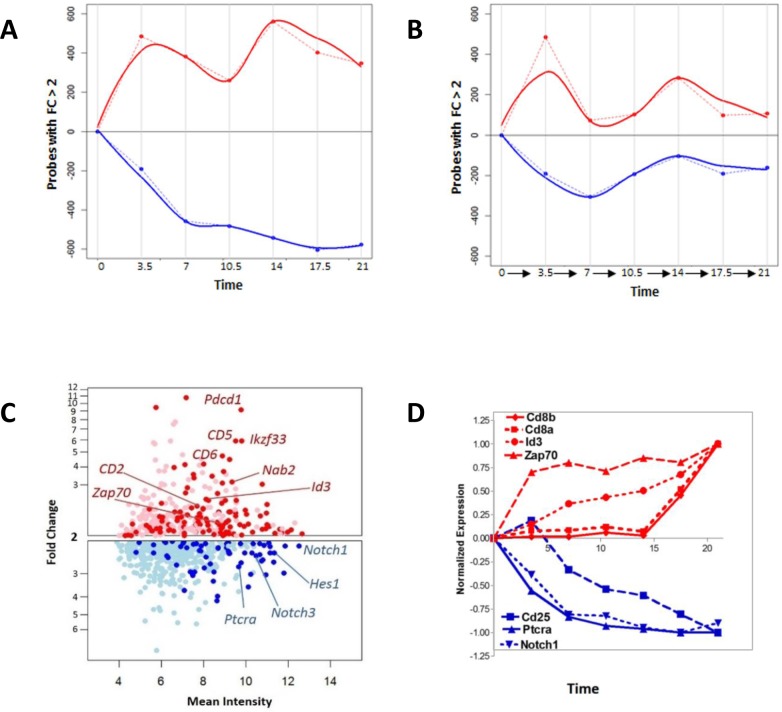
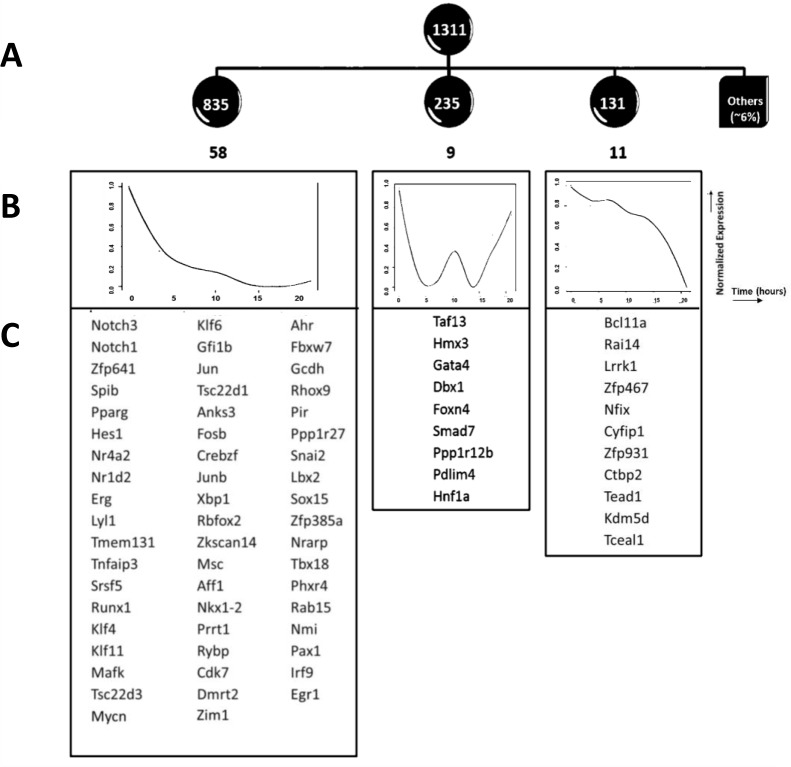
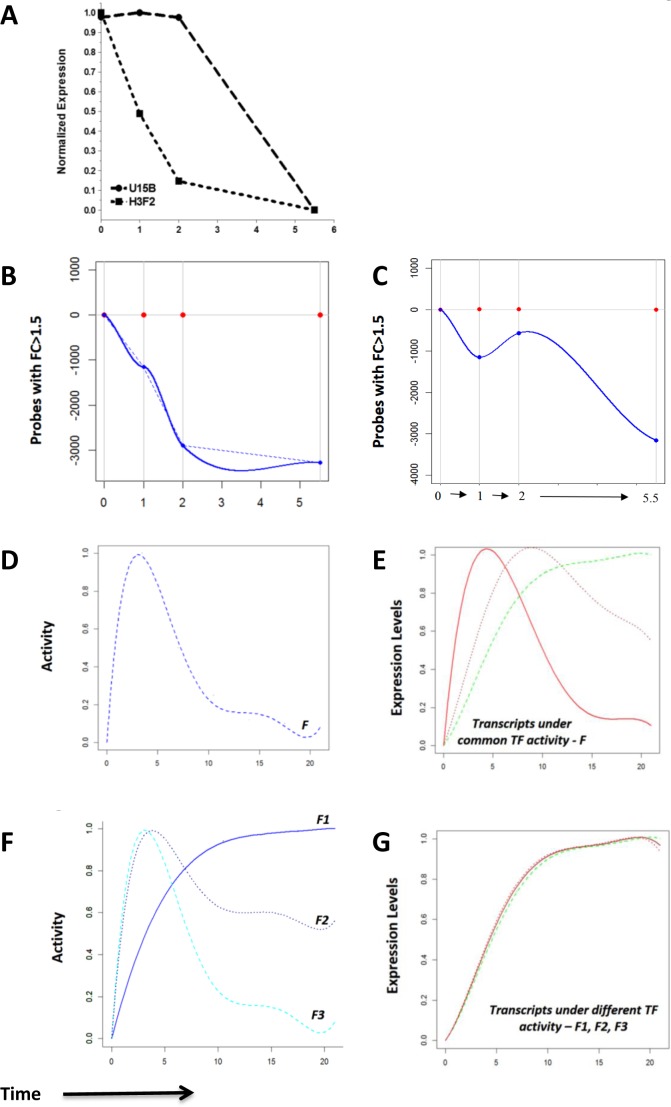
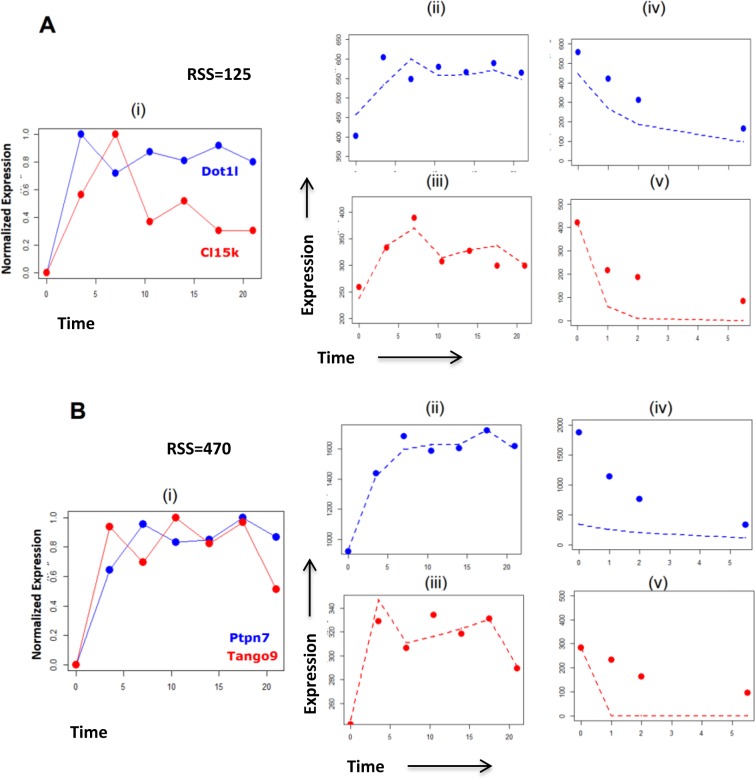
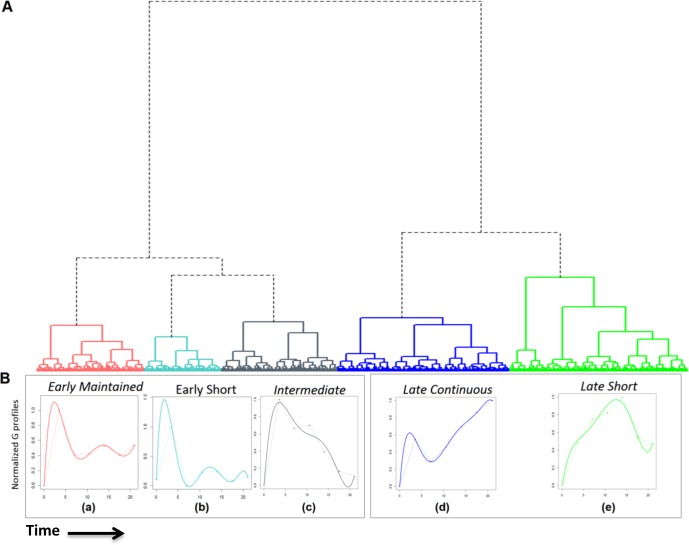
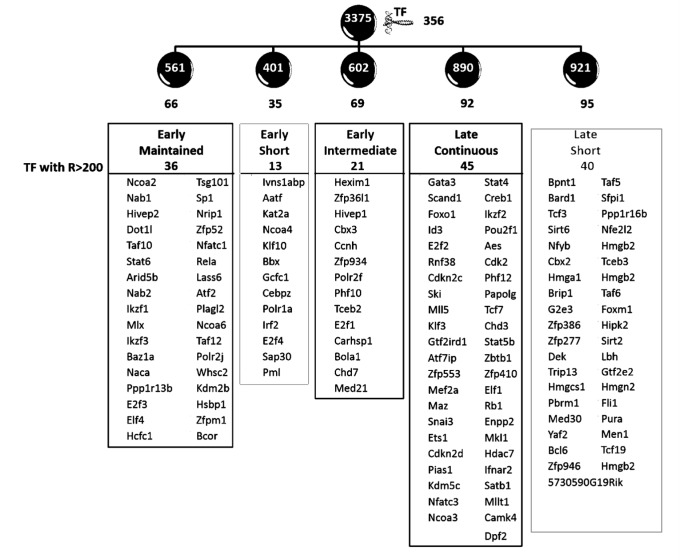
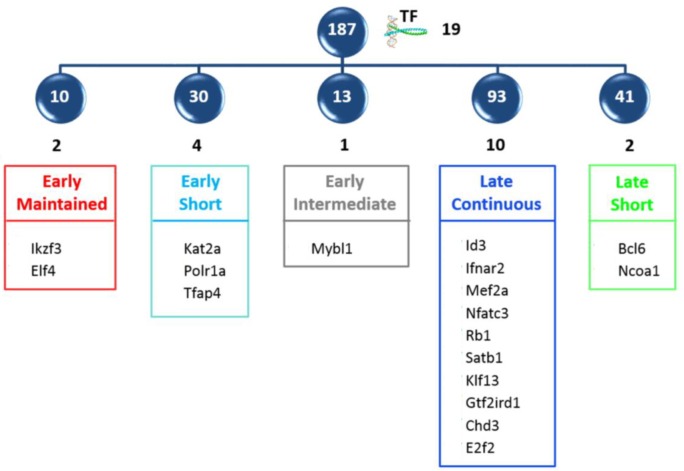
Developing thymocytes require pre-TCR signalling to differentiate from CD4-CD8- double negative to CD4+CD8+ double positive cell. Here we followed the transcriptional response to pre-TCR signalling in a synchronised population of differentiating double negative thymocytes. This time series analysis revealed a complex transcriptional response, in which thousands of genes were up and down-regulated before changes in cell surface phenotype were detected. Genome-wide measurement of RNA degradation of individual genes showed great heterogeneity in the rate of degradation between different genes. We therefore used time course expression and degradation data and a genome wide transcriptional modelling (GWTM) strategy to model the transcriptional response of genes up-regulated on pre-TCR signal transduction. This analysis revealed five major temporally distinct transcriptional activities that up regulate transcription through time, whereas down-regulation of expression occurred in three waves. Our model thus placed known regulators in a temporal perspective, and in addition identified novel candidate regulators of thymocyte differentiation.
Keywords: DP; Immune response; Immunity; Immunology Section; foetal thymic organ cultures; genome wide transcriptional modelling; pre-TCR; thymus.
Conflict of interest statement
The authors have no conflicts of interest.
Figures
References
-
- Koch U, Radtke F. Mechanisms of T cell development and transformation. Annual review of cell and developmental biology. 2011;27:539–562. - PubMed
-
- Shah DK, Zuniga-Pflucker JC. An Overview of the Intrathymic Intricacies of T Cell Development. Journal of Immunology. 2014;192:4017–4023. - PubMed
-
- Godfrey DI, Kennedy J, Suda T, Zlotnik A. A developmental pathway involving four phenotypically and functionally distinct subsets of CD3−CD4−CD8− triplenegative adult mouse thymocytes defined by CD44 and CD25 expression. Journal of immunology. 1993;150:4244–4252. - PubMed
-
- Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, Aguet M. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 1999;10:547–558. - PubMed
-
- Pui JC, Allman D, Xu L, DeRocco S, Karnell FG, Bakkour S, Lee JY, Kadesch T, Hardy RR, Aster JC, Pear WS. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity. 1999;11:299–308. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
