

Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis
- PMID: 26416663
- PMCID: PMC4587849
- DOI: 10.1186/s13073-015-0220-9
Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis
Abstract
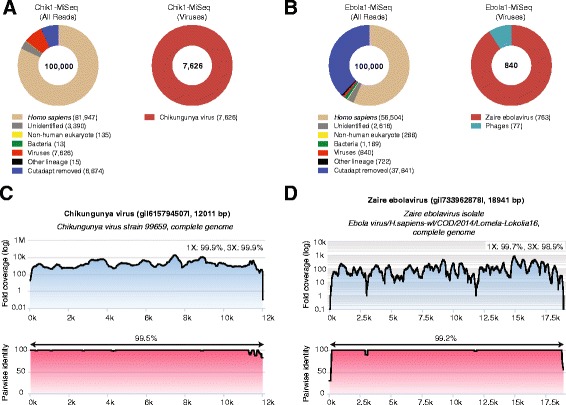
We report unbiased metagenomic detection of chikungunya virus (CHIKV), Ebola virus (EBOV), and hepatitis C virus (HCV) from four human blood samples by MinION nanopore sequencing coupled to a newly developed, web-based pipeline for real-time bioinformatics analysis on a computational server or laptop (MetaPORE). At titers ranging from 10(7)-10(8) copies per milliliter, reads to EBOV from two patients with acute hemorrhagic fever and CHIKV from an asymptomatic blood donor were detected within 4 to 10 min of data acquisition, while lower titer HCV virus (1 × 10(5) copies per milliliter) was detected within 40 min. Analysis of mapped nanopore reads alone, despite an average individual error rate of 24 % (range 8-49 %), permitted identification of the correct viral strain in all four isolates, and 90 % of the genome of CHIKV was recovered with 97-99 % accuracy. Using nanopore sequencing, metagenomic detection of viral pathogens directly from clinical samples was performed within an unprecedented <6 hr sample-to-answer turnaround time, and in a timeframe amenable to actionable clinical and public health diagnostics.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
