

A novel locus of resistance to severe malaria in a region of ancient balancing selection
- PMID: 26416757
- PMCID: PMC4629224
- DOI: 10.1038/nature15390
A novel locus of resistance to severe malaria in a region of ancient balancing selection
Abstract
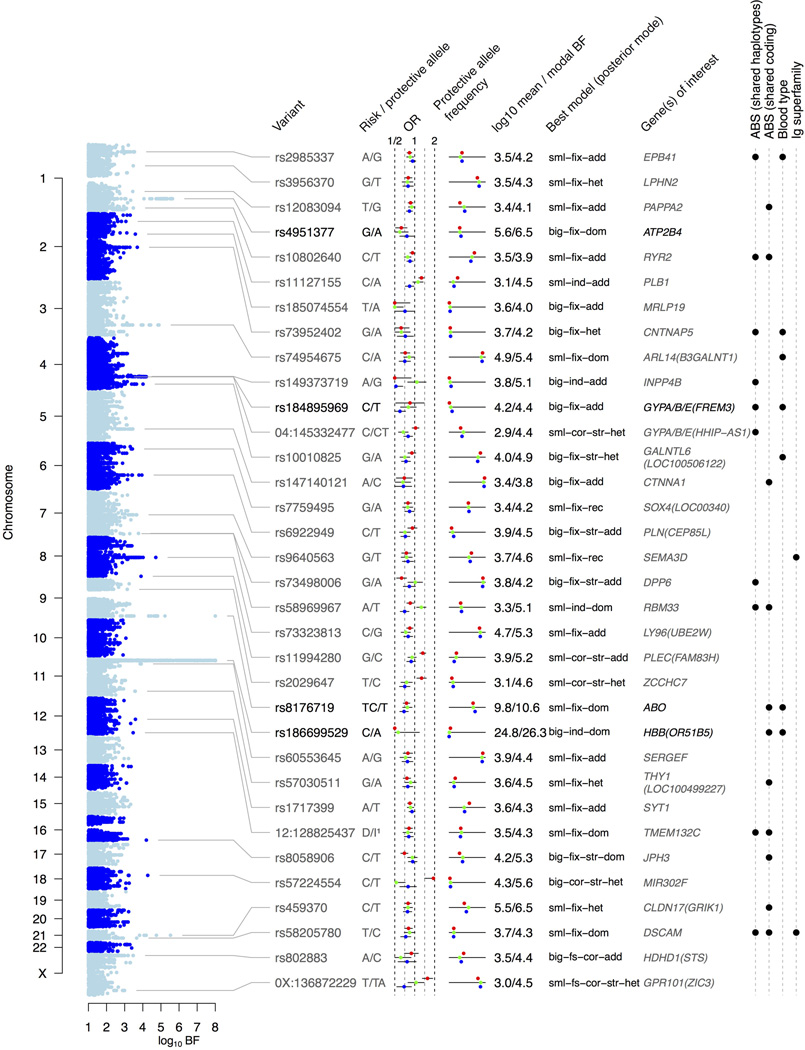
The high prevalence of sickle haemoglobin in Africa shows that malaria has been a major force for human evolutionary selection, but surprisingly few other polymorphisms have been proven to confer resistance to malaria in large epidemiological studies. To address this problem, we conducted a multi-centre genome-wide association study (GWAS) of life-threatening Plasmodium falciparum infection (severe malaria) in over 11,000 African children, with replication data in a further 14,000 individuals. Here we report a novel malaria resistance locus close to a cluster of genes encoding glycophorins that are receptors for erythrocyte invasion by P. falciparum. We identify a haplotype at this locus that provides 33% protection against severe malaria (odds ratio = 0.67, 95% confidence interval = 0.60-0.76, P value = 9.5 × 10(-11)) and is linked to polymorphisms that have previously been shown to have features of ancient balancing selection, on the basis of haplotype sharing between humans and chimpanzees. Taken together with previous observations on the malaria-protective role of blood group O, these data reveal that two of the strongest GWAS signals for severe malaria lie in or close to genes encoding the glycosylated surface coat of the erythrocyte cell membrane, both within regions of the genome where it appears that evolution has maintained diversity for millions of years. These findings provide new insights into the host-parasite interactions that are critical in determining the outcome of malaria infection.
Figures












Comment in
-
Glycophorins, Blood Groups, and Protection from Severe Malaria.Trends Parasitol. 2016 Jan;32(1):5-7. doi: 10.1016/j.pt.2015.11.006. Epub 2015 Nov 21. Trends Parasitol. 2016. PMID: 26613665 Free PMC article.
References
-
- Timmann C, et al. Genome-wide association study indicates two novel resistance loci for severe malaria. Nature. 2012 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 096527/WT_/Wellcome Trust/United Kingdom
- 091758/WT_/Wellcome Trust/United Kingdom
- G0600230/MRC_/Medical Research Council/United Kingdom
- 097364/Z/11/Z/WT_/Wellcome Trust/United Kingdom
- G0600718/MRC_/Medical Research Council/United Kingdom
- N01 AI085346/AI/NIAID NIH HHS/United States
- 084538/WT_/Wellcome Trust/United Kingdom
- U19AI065683/AI/NIAID NIH HHS/United States
- 087285/WT_/Wellcome Trust/United Kingdom
- 090770/WT_/Wellcome Trust/United Kingdom
- 098051/WT_/Wellcome Trust/United Kingdom
- MC_UP_A900_1118/MRC_/Medical Research Council/United Kingdom
- D43TW001589/TW/FIC NIH HHS/United States
- G9901439/MRC_/Medical Research Council/United Kingdom
- 075491/Z/04/WT_/Wellcome Trust/United Kingdom
- 091758/Z/10/Z/WT_/Wellcome Trust/United Kingdom
- 090770/Z/09/Z/WT_/Wellcome Trust/United Kingdom
- MR/M006212/1/MRC_/Medical Research Council/United Kingdom
- WT077383/Z/05/Z/WT_/Wellcome Trust/United Kingdom
- G19/9/MRC_/Medical Research Council/United Kingdom
- 077012/Z/05/Z/WT_/Wellcome Trust/United Kingdom
- U19 AI065683/AI/NIAID NIH HHS/United States
- 090532/Z/09/Z/WT_/Wellcome Trust/United Kingdom
- 076934/Z/05/Z/WT_/Wellcome Trust/United Kingdom
- D43 TW001589/TW/FIC NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
