

mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis
- PMID: 26416965
- PMCID: PMC4586745
- DOI: 10.1083/jcb.201503066
mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis
Abstract
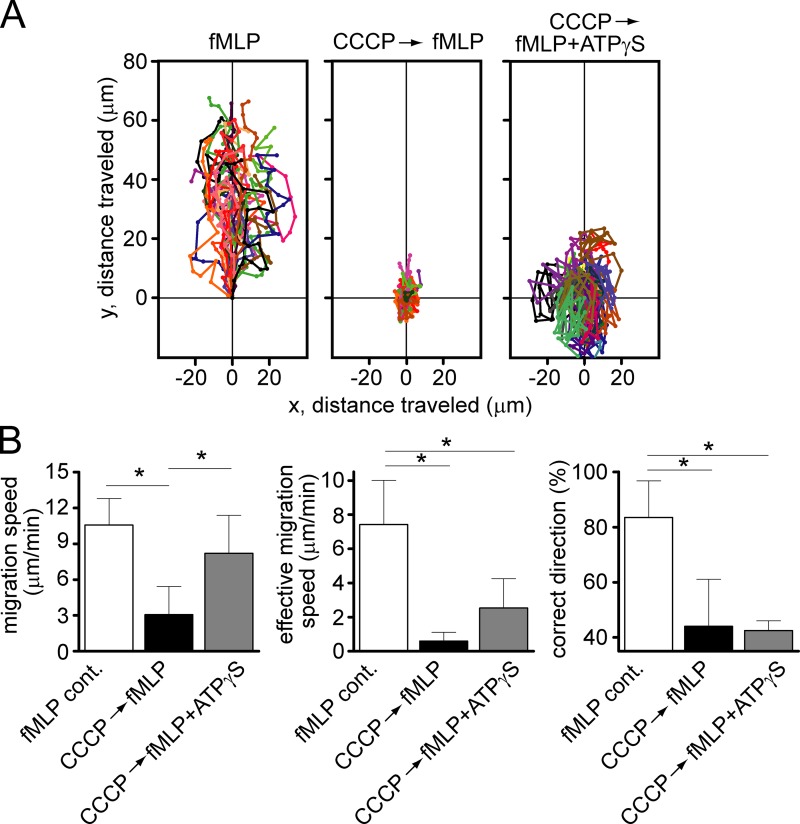
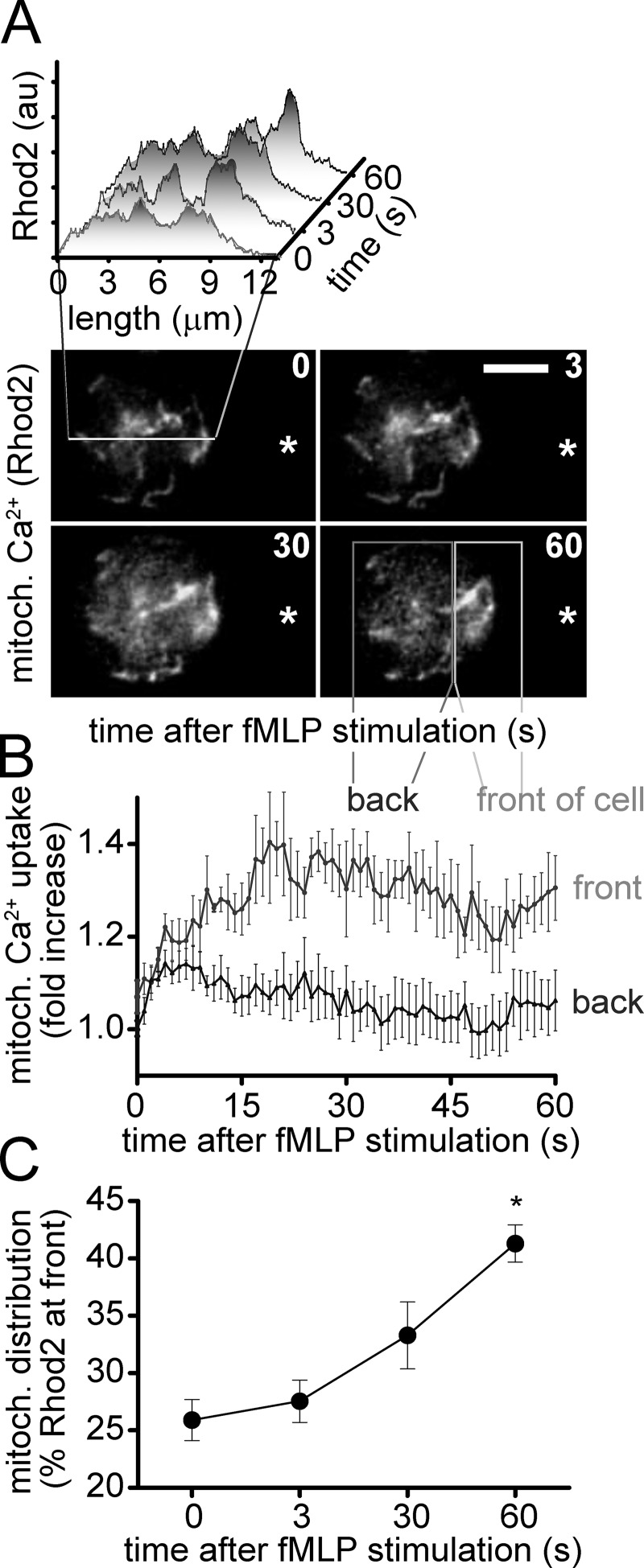
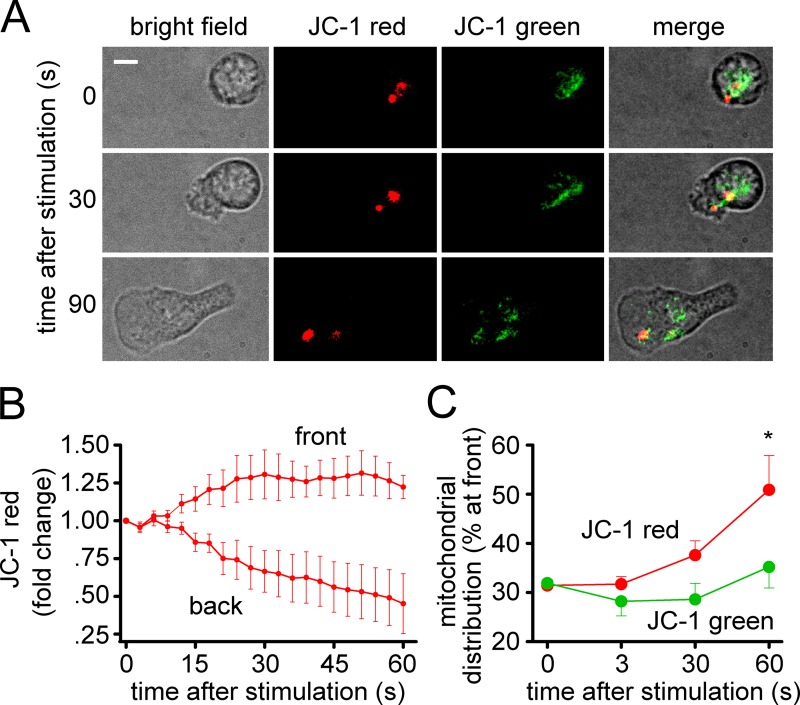
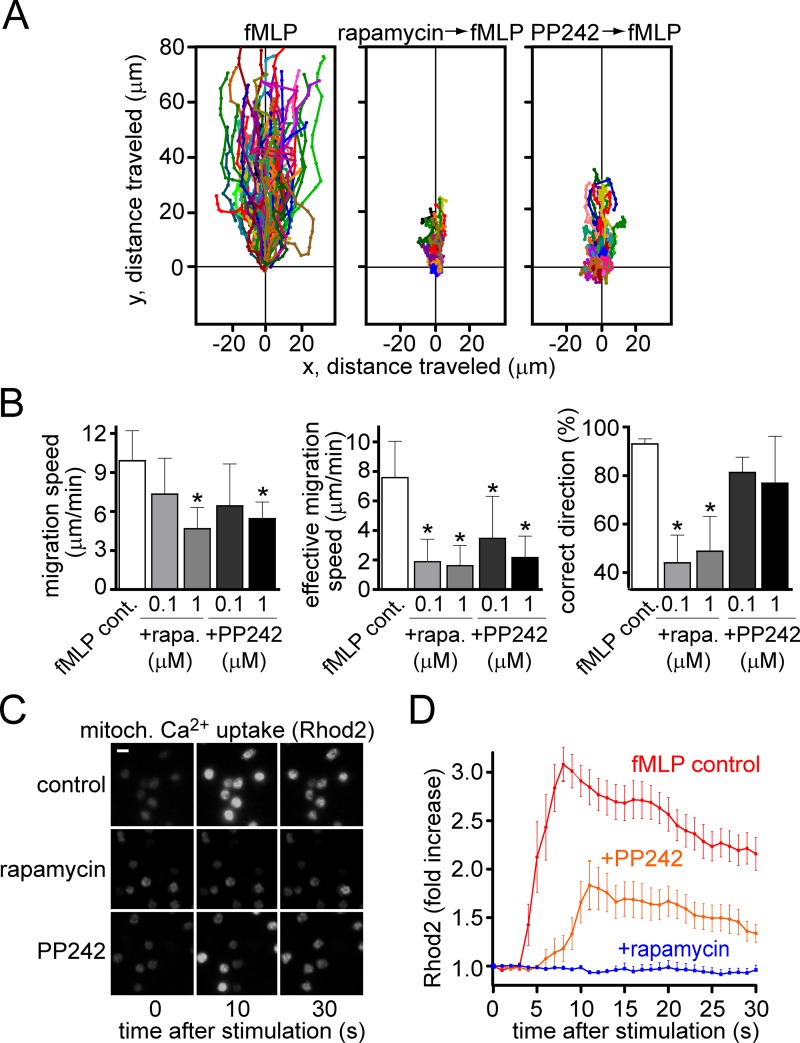
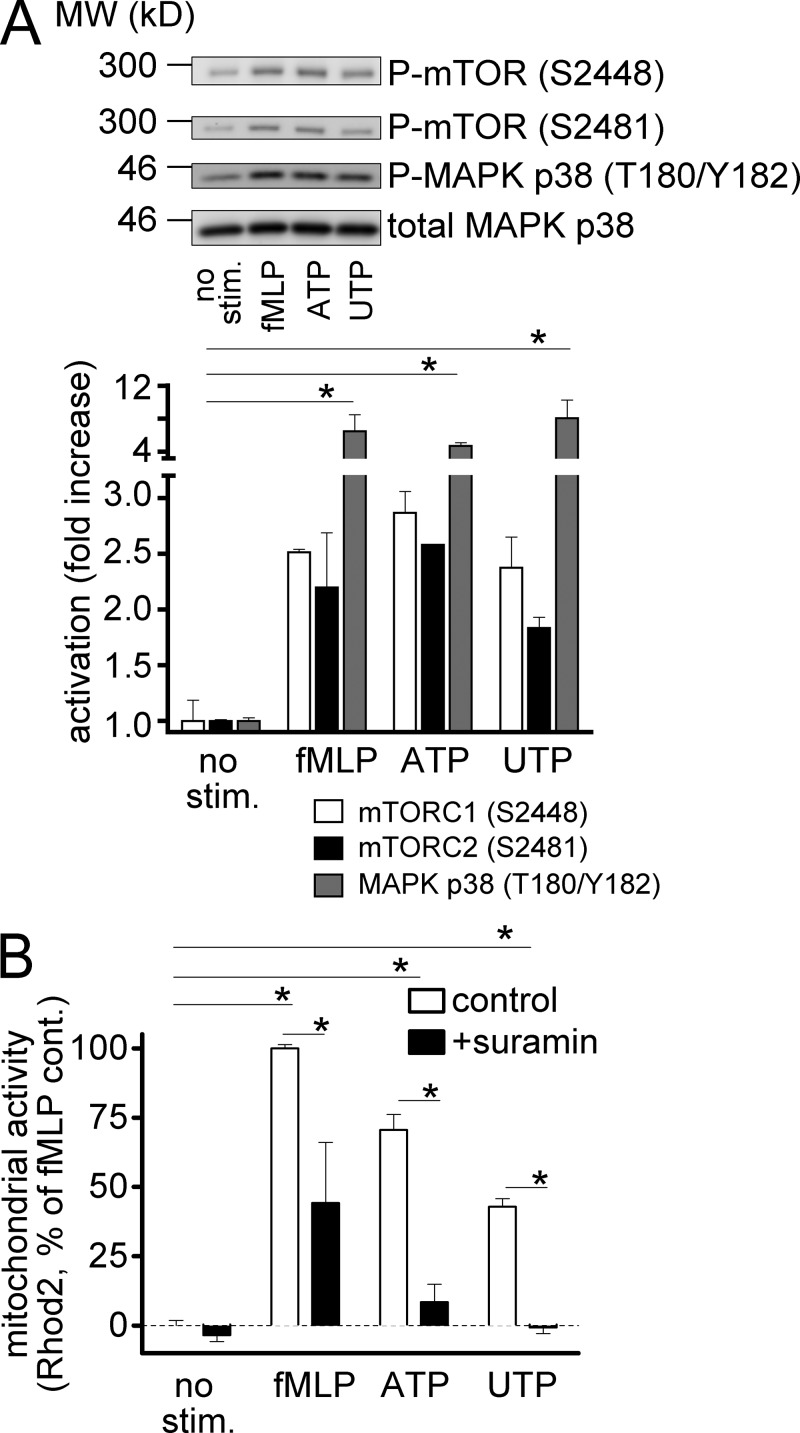
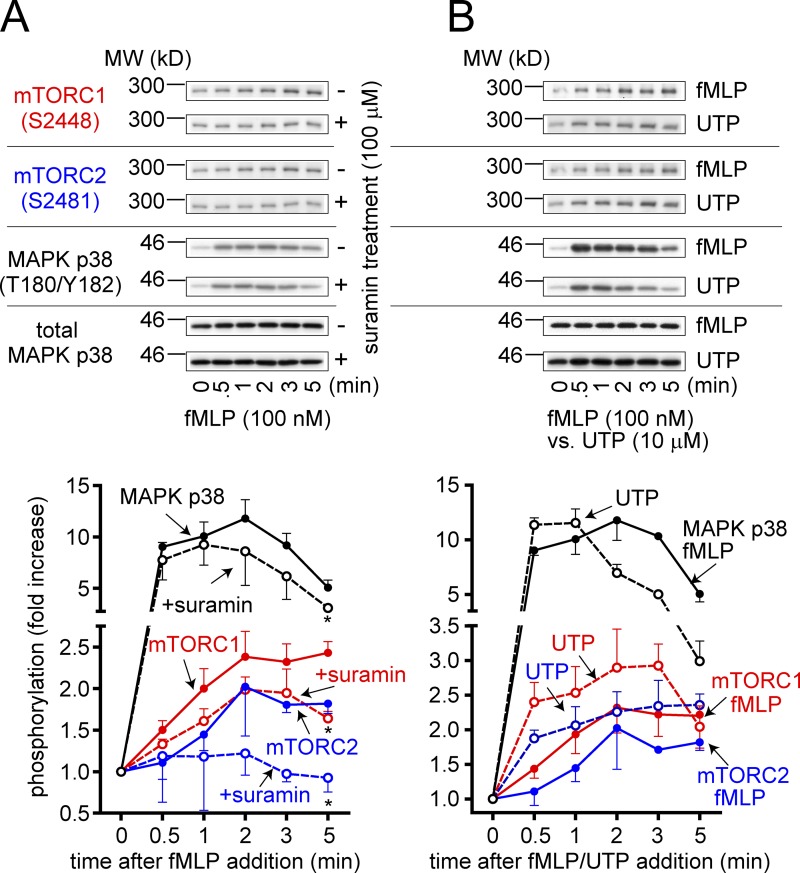
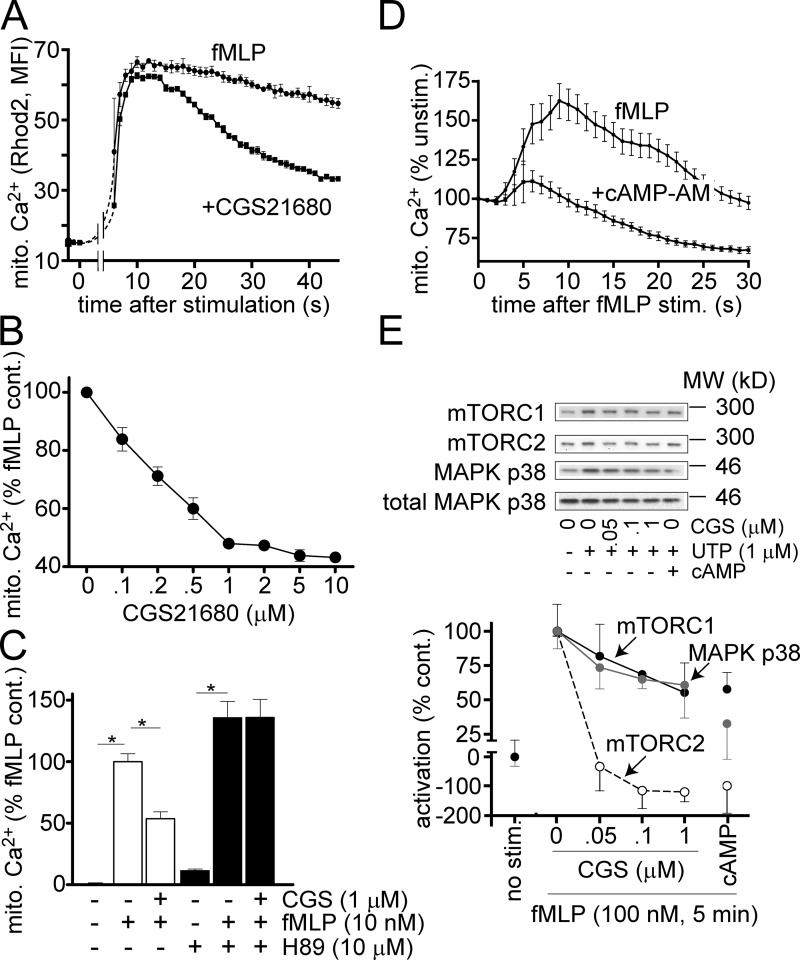
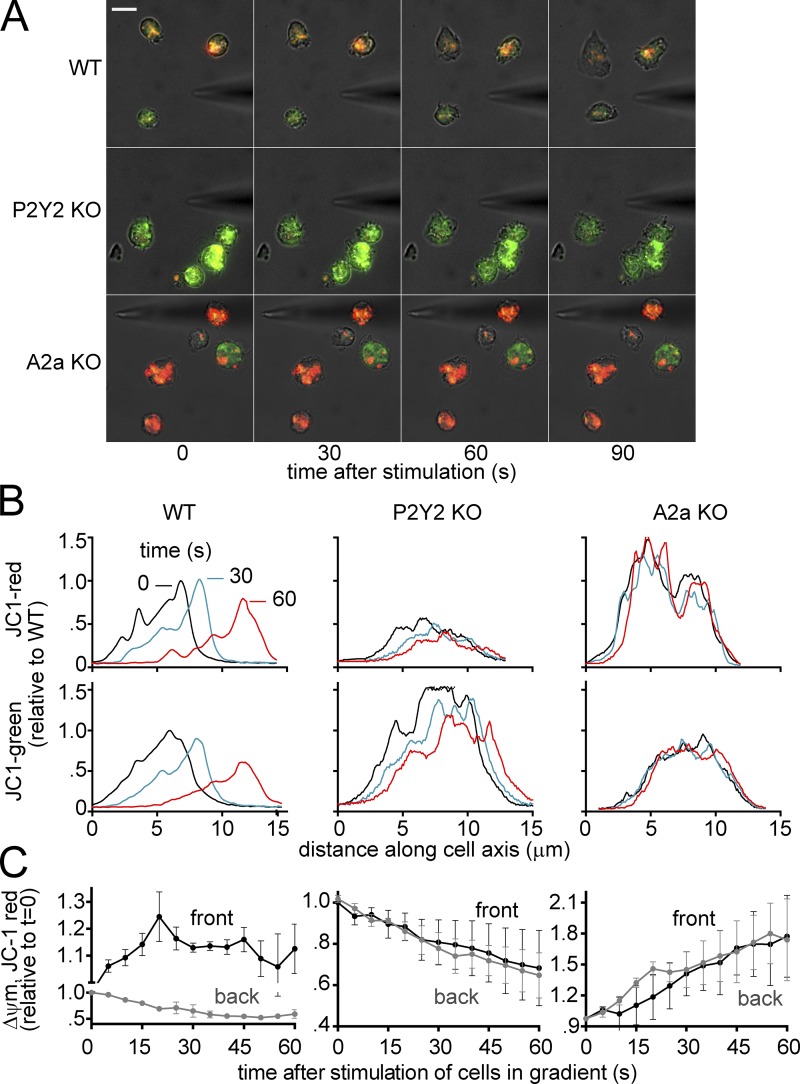
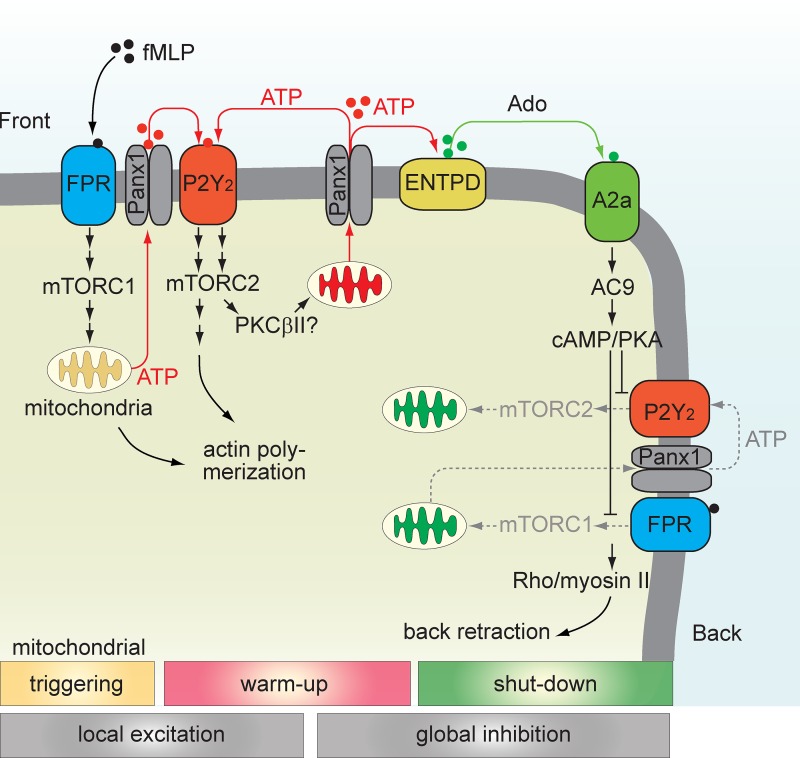
Neutrophils use chemotaxis to locate invading bacteria. Adenosine triphosphate (ATP) release and autocrine purinergic signaling via P2Y2 receptors at the front and A2a receptors at the back of cells regulate chemotaxis. Here, we examined the intracellular mechanisms that control these opposing signaling mechanisms. We found that mitochondria deliver ATP that stimulates P2Y2 receptors in response to chemotactic cues, and that P2Y2 receptors promote mTOR signaling, which augments mitochondrial activity near the front of cells. Blocking mTOR signaling with rapamycin or PP242 or mitochondrial ATP production (e.g., with CCCP) reduced mitochondrial Ca(2+) uptake and membrane potential, and impaired cellular ATP release and neutrophil chemotaxis. Autocrine stimulation of A2a receptors causes cyclic adenosine monophosphate accumulation at the back of cells, which inhibits mTOR signaling and mitochondrial activity, resulting in uropod retraction. We conclude that mitochondrial, purinergic, and mTOR signaling regulates neutrophil chemotaxis and may be a pharmacological target in inflammatory diseases.
© 2015 Bao et al.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
