

TPM3 deletions cause a hypercontractile congenital muscle stiffness phenotype
- PMID: 26418456
- PMCID: PMC5154623
- DOI: 10.1002/ana.24535
TPM3 deletions cause a hypercontractile congenital muscle stiffness phenotype
Abstract
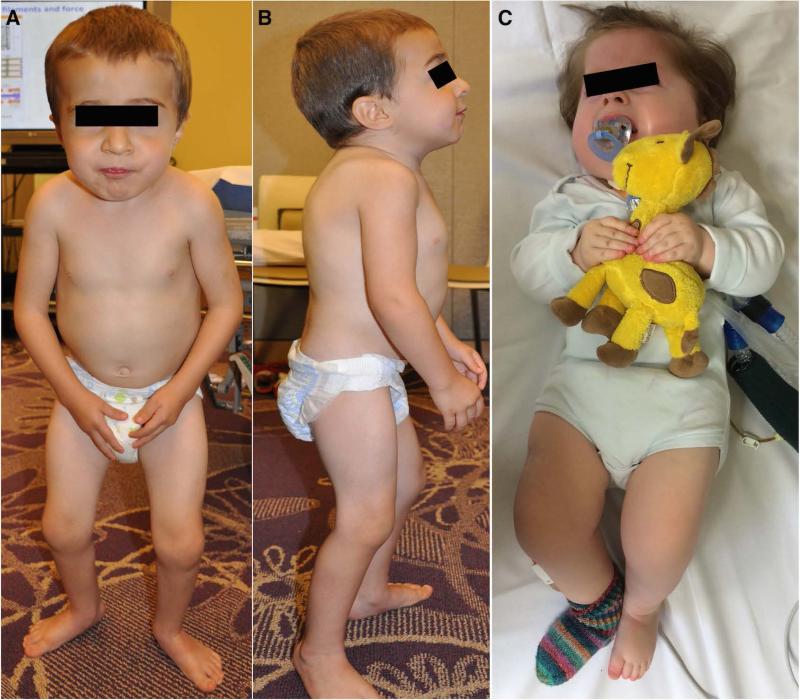
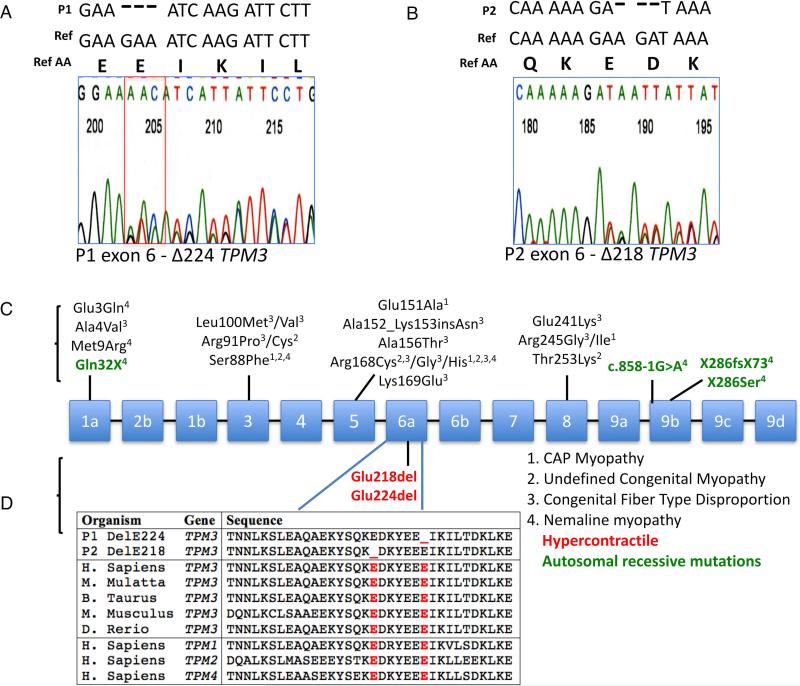
Objective: Mutations in TPM3, encoding Tpm3.12, cause a clinically and histopathologically diverse group of myopathies characterized by muscle weakness. We report two patients with novel de novo Tpm3.12 single glutamic acid deletions at positions ΔE218 and ΔE224, resulting in a significant hypercontractile phenotype with congenital muscle stiffness, rather than weakness, and respiratory failure in one patient.
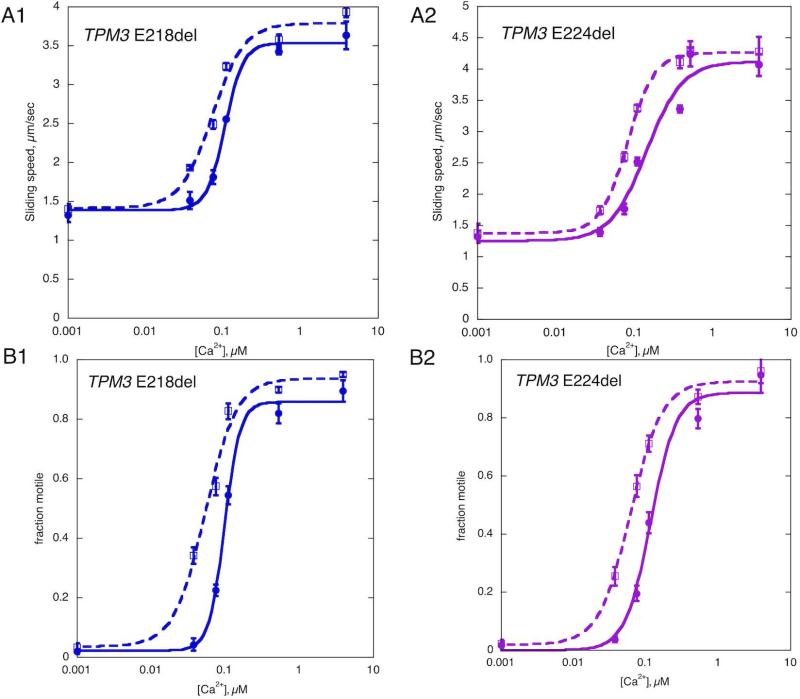
Methods: The effect of the Tpm3.12 deletions on the contractile properties in dissected patient myofibers was measured. We used quantitative in vitro motility assay to measure Ca(2+) sensitivity of thin filaments reconstituted with recombinant Tpm3.12 ΔE218 and ΔE224.
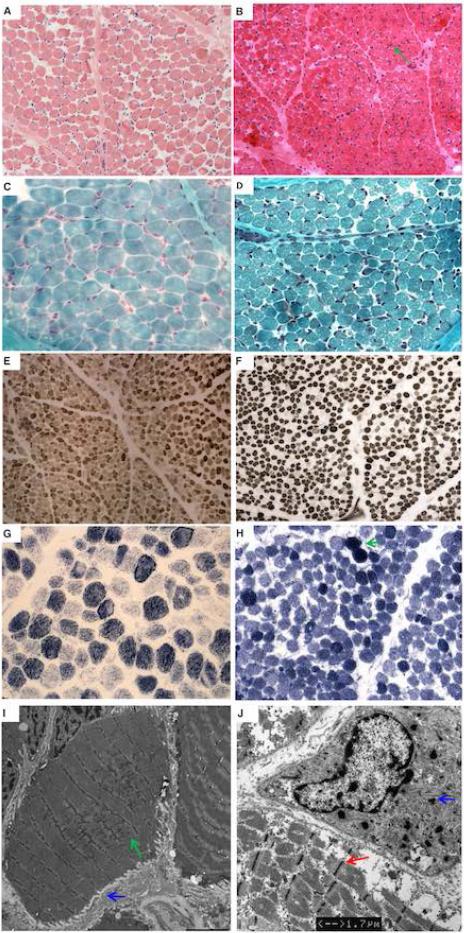
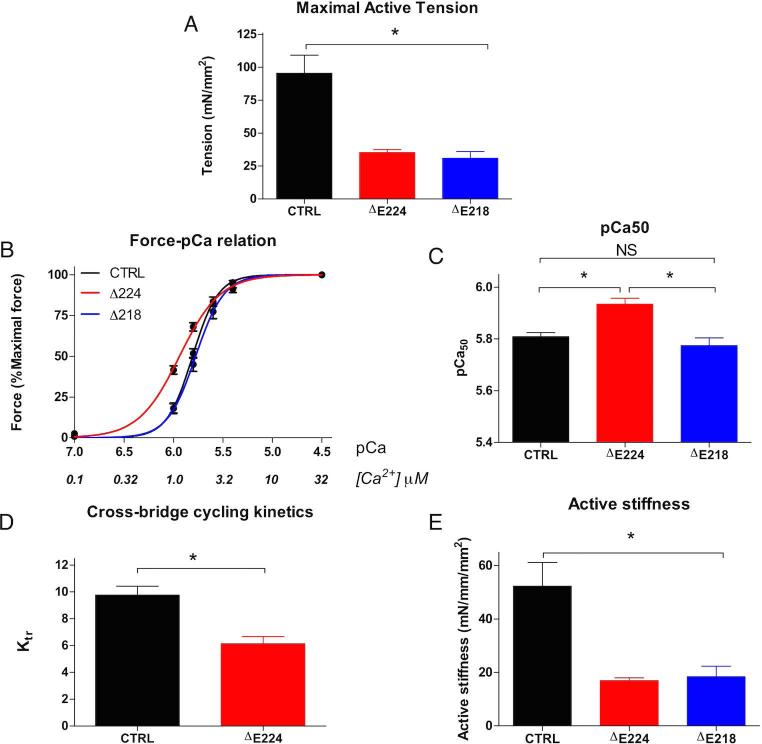
Results: Contractility studies on permeabilized myofibers demonstrated reduced maximal active tension from both patients with increased Ca(2+) sensitivity and altered cross-bridge cycling kinetics in ΔE224 fibers. In vitro motility studies showed a two-fold increase in Ca(2+) sensitivity of the fraction of filaments motile and the filament sliding velocity concentrations for both mutations.
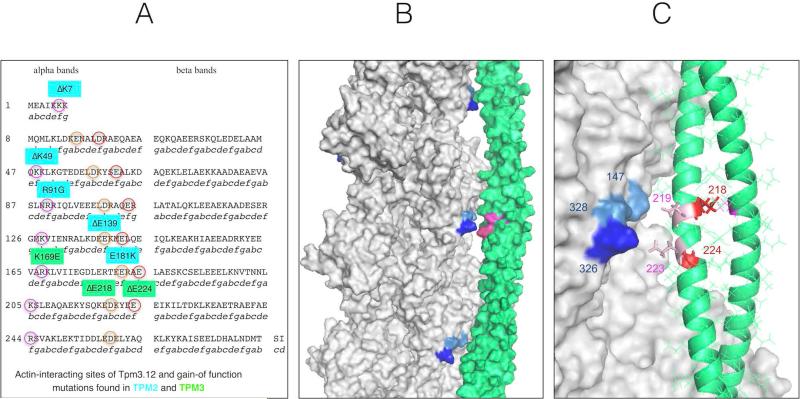
Interpretation: These data indicate that Tpm3.12 deletions ΔE218 and ΔE224 result in increased Ca(2+) sensitivity of the troponin-tropomyosin complex, resulting in abnormally active interaction of the actin and myosin complex. Both mutations are located in the charged motifs of the actin-binding residues of tropomyosin 3, thus disrupting the electrostatic interactions that facilitate accurate tropomyosin binding with actin necessary to prevent the on-state. The mutations destabilize the off-state and result in excessively sensitized excitation-contraction coupling of the contractile apparatus. This work expands the phenotypic spectrum of TPM3-related disease and provides insights into the pathophysiological mechanisms of the actin-tropomyosin complex.
© 2015 American Neurological Association.
Figures
References
-
- Perry SV. Vertebrate tropomyosin: Distribution, properties and function. J. Muscle Res. Cell Motil. 2001;22:5–49. - PubMed
-
- De Paula AM, Franques J, Fernandez C, et al. A TPM3 mutation causing cap myopathy. Neuromuscul. Disord. 2009;19:685–688. - PubMed
-
- Ohlsson M, Fidzianska A, Tajsharghi H, Oldfors A. TPM3 mutation in one of the original cases of cap disease. Neurology. 2009;72(22):1961–3. - PubMed
-
- Schreckenbach T, Schröder JM, Voit T, et al. Novel TPM3 mutation in a family with cap myopathy and review of the literature. Neuromuscul. Disord. 2014;24(2):117–24. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
