

Advancing Urinary Protein Biomarker Discovery by Data-Independent Acquisition on a Quadrupole-Orbitrap Mass Spectrometer
- PMID: 26423119
- PMCID: PMC4993212
- DOI: 10.1021/acs.jproteome.5b00826
Advancing Urinary Protein Biomarker Discovery by Data-Independent Acquisition on a Quadrupole-Orbitrap Mass Spectrometer
Abstract
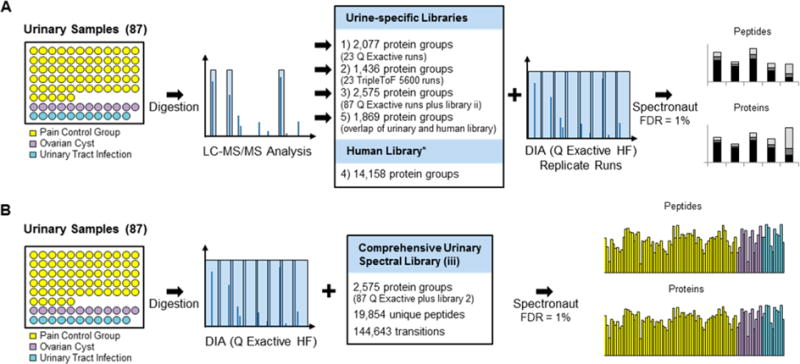
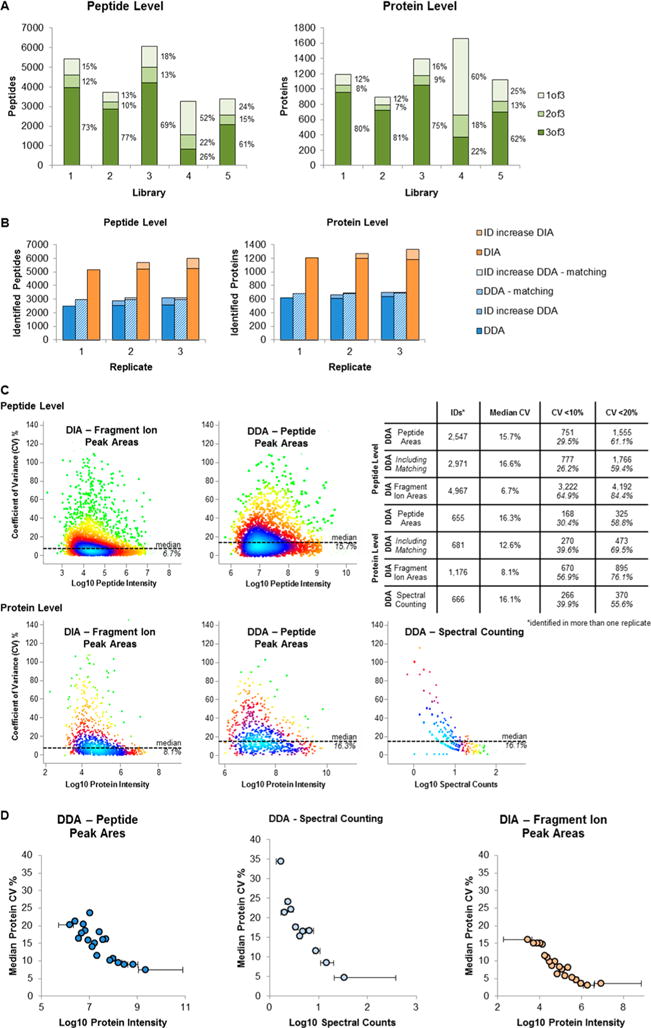
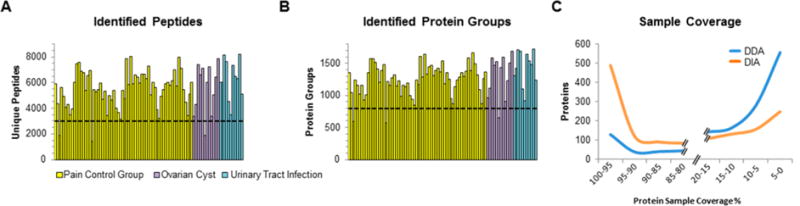
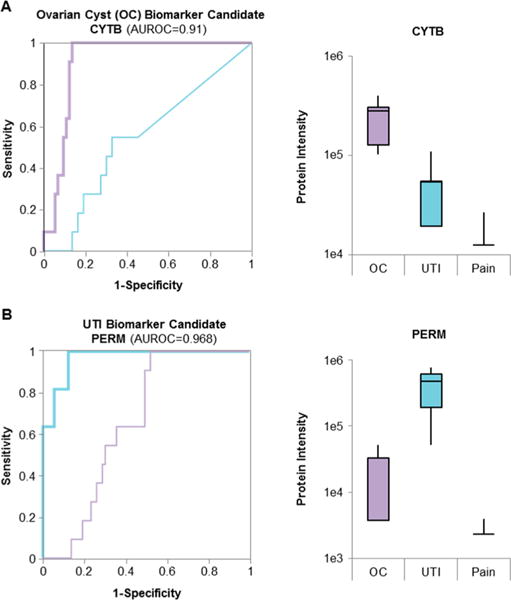
The promises of data-independent acquisition (DIA) strategies are a comprehensive and reproducible digital qualitative and quantitative record of the proteins present in a sample. We developed a fast and robust DIA method for comprehensive mapping of the urinary proteome that enables large scale urine proteomics studies. Compared to a data-dependent acquisition (DDA) experiments, our DIA assay doubled the number of identified peptides and proteins per sample at half the coefficients of variation observed for DDA data (DIA = ∼8%; DDA = ∼16%). We also tested different spectral libraries and their effects on overall protein and peptide identifications and their reproducibilities, which provided clear evidence that sample type-specific spectral libraries are preferred for reliable data analysis. To show applicability for biomarker discovery experiments, we analyzed a sample set of 87 urine samples from children seen in the emergency department with abdominal pain. The whole set was analyzed with high proteome coverage (∼1300 proteins/sample) in less than 4 days. The data set revealed excellent biomarker candidates for ovarian cyst and urinary tract infection. The improved throughput and quantitative performance of our optimized DIA workflow allow for the efficient simultaneous discovery and verification of biomarker candidates without the requirement for an early bias toward selected proteins.
Keywords: DIA; QE; biomarker discovery; spectral library; urine proteomics.
Conflict of interest statement
Notes The authors declare no competing financial interest.
Figures
References
-
- Wu J, Chen YD, Gu W. Urinary proteomics as a novel tool for biomarker discovery in kidney diseases. J Zhejiang Univ Sci B. 2010;11:227–37. - PMC - PubMed
- Lopez-Giacoman S, Madero M. Biomarkers in chronic kidney disease from kidney function to kidney damage. World J Nephrol. 2015;4:57–73. - PMC - PubMed
- Pedroza-Diaz J, Rothlisberger S. Advances in urinary protein biomarkers for urogenital non-urogenital pathologies. Biochem Med (Zagreb) 2015;25:22–35. - PMC - PubMed
- Mischak H, Delles C, Vlahou A, Vanholder R. Proteomic biomarkers in kidney disease: issues in development implementation. Nat Rev Nephrol. 2015;11:221–32. - PubMed
-
- Kentsis A, Shulman A, Ahmed S, Brennan E, Monuteaux MC, Lee YH, Lipsett S, Paulo JA, Dedeoglu F, Fuhlbrigge R, Bachur R, Bradwin G, Arditi M, Sundel RP, Newburger JW, Steen H, Kim S. Urine proteomics for discovery of improved diagnostic markers of Kawasaki disease. EMBO molecular medicine. 2013;5:210–20. - PMC - PubMed
-
- Zimmerli LU, Schiffer E, Zurbig P, Good DM, Kellmann M, Mouls L, Pitt AR, Coon JJ, Schmieder RE, Peter KH, Mischak H, Kolch W, Delles C, Dominiczak AF. Urinary proteomic biomarkers in coronary artery disease. Mol Cell Proteomics. 2007;7:290–8. - PubMed
-
- Adeola HA, Soares NC, Paccez JD, Kaestner L, Blackburn JM, Zerbini LF. Discovery of novel candidate urinary protein biomarkers for prostate cancer in a multi-ethnic cohort of South African patients via label-free mass spectrometry. Proteomics: Clin Appl. 2015;9:597–609. - PubMed
-
- Kentsis A, Lin YY, Kurek K, Calicchio M, Wang YY, Monigatti F, Campagne F, Lee R, Horwitz B, Steen H, Bachur R. Discovery validation of urine markers of acute pediatric appendicitis using high-accuracy mass spectrometry. Annals of emergency medicine. 2010;55:62–70. - PMC - PubMed
- Kentsis A, Ahmed S, Kurek K, Brennan E, Bradwin G, Steen H, Bachur R. Detection diagnostic value of urine leucine-rich alpha-2-glycoprotein in children with suspected acute appendicitis. Annals of emergency medicine. 2012;60:78–83. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
