

PSR: polymorphic SSR retrieval
- PMID: 26428628
- PMCID: PMC4591729
- DOI: 10.1186/s13104-015-1474-4
PSR: polymorphic SSR retrieval
Abstract
Background: With the advent of high-throughput sequencing technologies large-scale identification of microsatellites became affordable and was especially directed to non-model species. By contrast, few efforts have been published toward the automatic identification of polymorphic microsatellites by exploiting sequence redundancy. Few tools for genotyping microsatellite repeats have been implemented so far that are able to manage huge amount of sequence data and handle the SAM/BAM file format. Most of them have been developed for and tested on human or model organisms with high quality reference genomes.
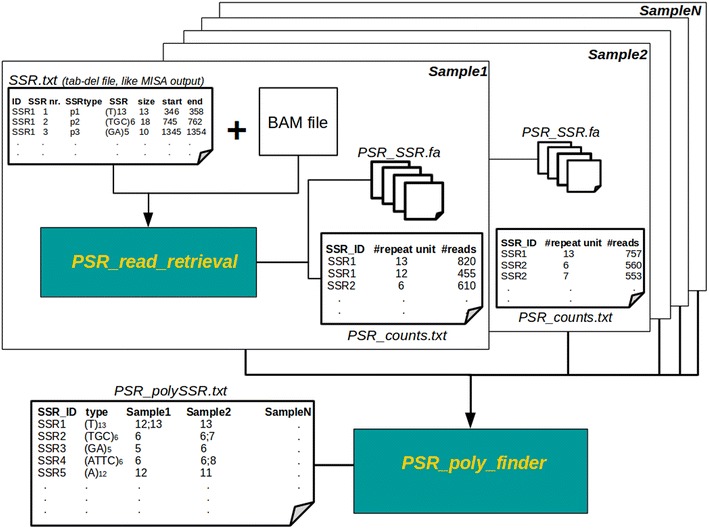
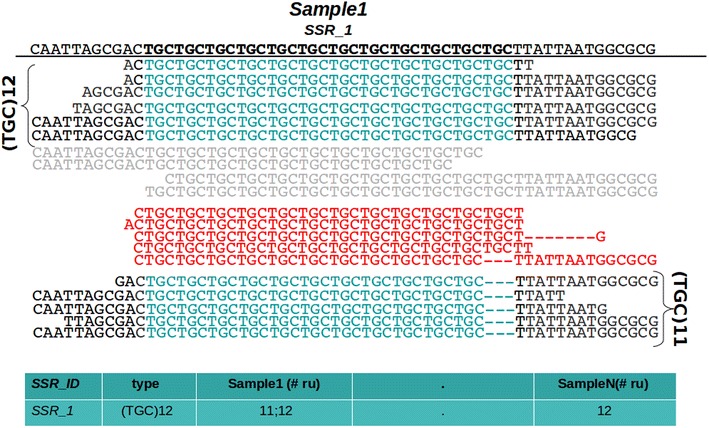
Results: In this note we describe polymorphic SSR retrieval (PSR), a read counter and simple sequence repeat (SSR) length polymorphism detection tool. It is written in Perl and was developed to identify length polymorphisms in perfect microsatellites exploiting next generation sequencing (NGS) data. PSR has been developed bearing in mind plant non-model species for which de novo transcriptome assembly is generally the first sequence resource available to be used for SSR-mining. PSR is divided into two modules: the read-counting module (PSR_read_retrieval) identifies all the reads that cover the full-length of perfect microsatellites; the comparative module (PSR_poly_finder) detects both heterozygous and homozygous alleles at each microsatellite locus across all genotypes under investigation. Two threshold values to call a length polymorphism and reduce the number of false positives can be defined by the user: the minimum number of reads overlapping the repetitive stretch and the minimum read depth. The first parameter determines if the microsatellite-containing sequence must be processed or not, while the second one is decisive for the identification of minor alleles. PSR was tested on two different case studies. The first study aims at the identification of polymorphic SSRs in a set of de novo assembled transcripts defined by RNA-sequencing of two different plant genotypes. The second research activity aims to investigate sequence variations within a collection of newly sequenced chloroplast genomes. In both the cases PSR results are in agreement with those obtained by capillary gel separation.
Conclusion: PSR has been specifically developed from the need to automate the gene-based and genome-wide identification of polymorphic microsatellites from NGS data. It overcomes the limits related to the existing and time-consuming efforts based on tools developed in the pre-NGS era.
Figures
References
-
- Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 2001;11(8):1441–1452. doi: 10.1101/gr.184001. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
