

Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes
- PMID: 26431796
- PMCID: PMC4651811
- DOI: 10.1016/j.taap.2015.09.022
Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes
Abstract
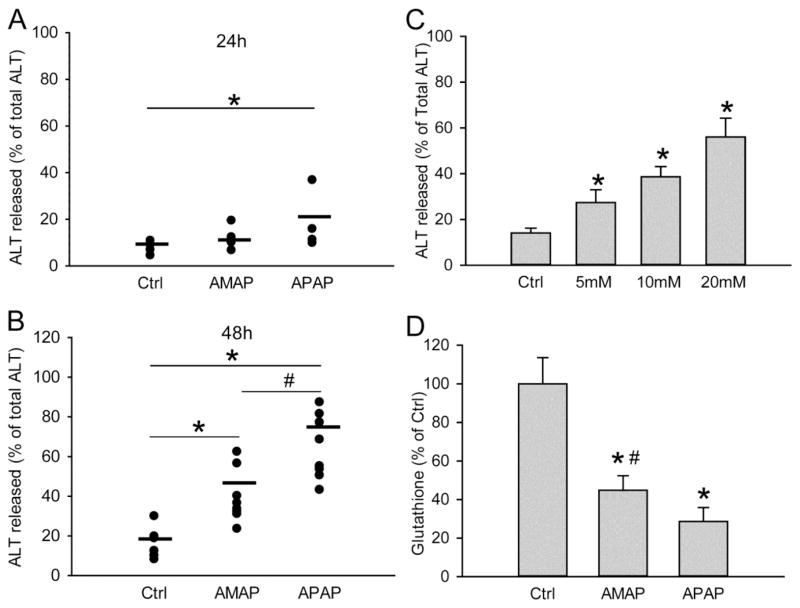
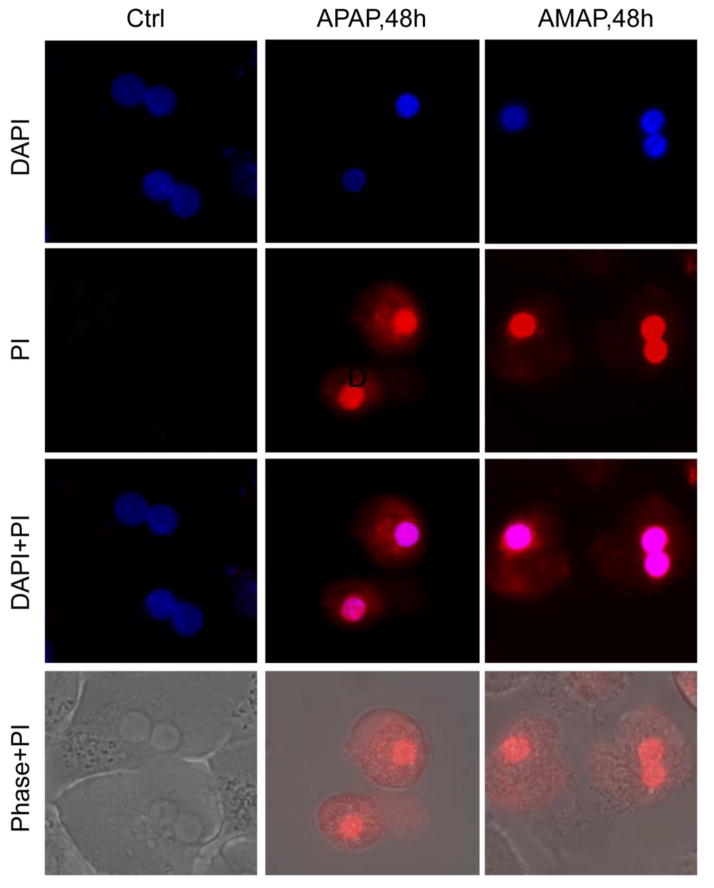
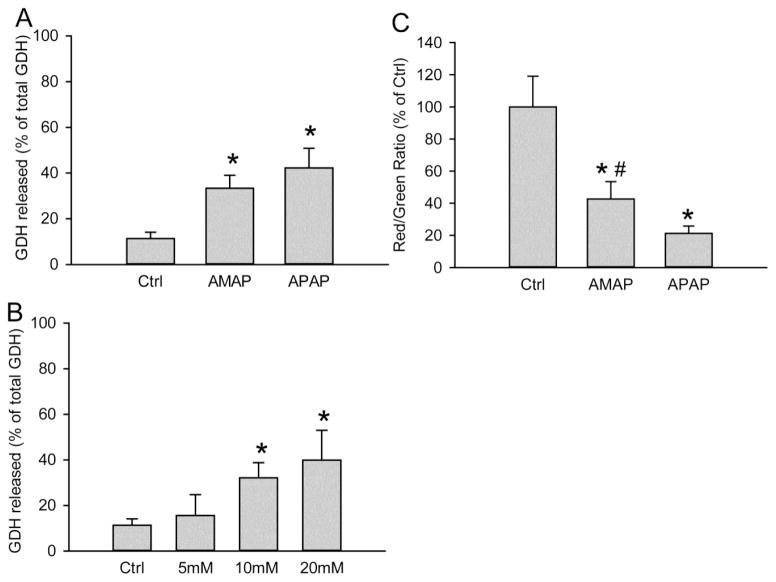
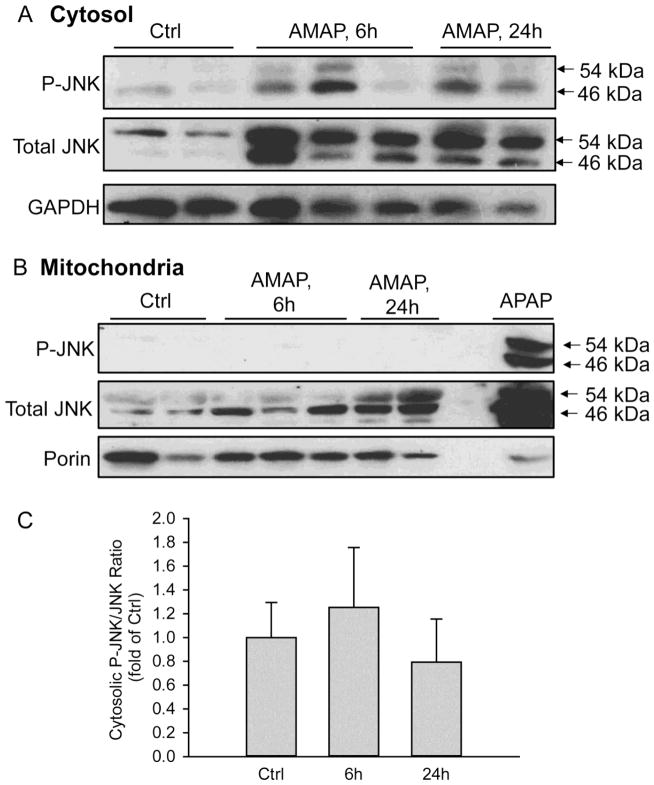
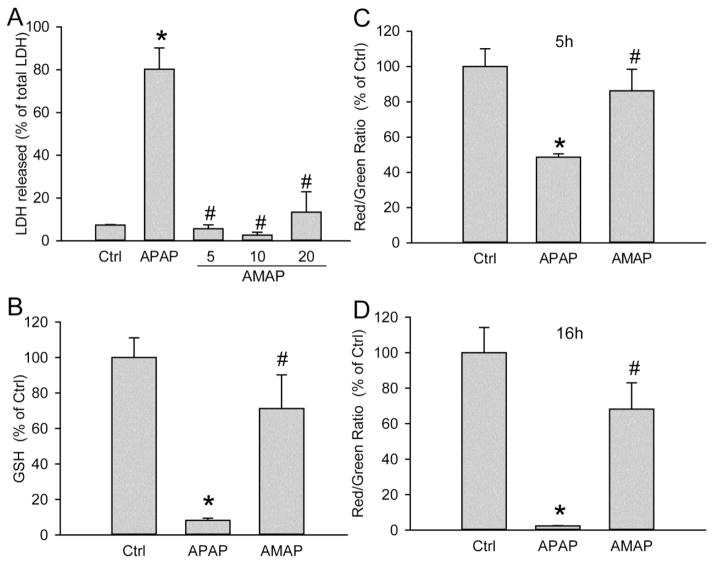
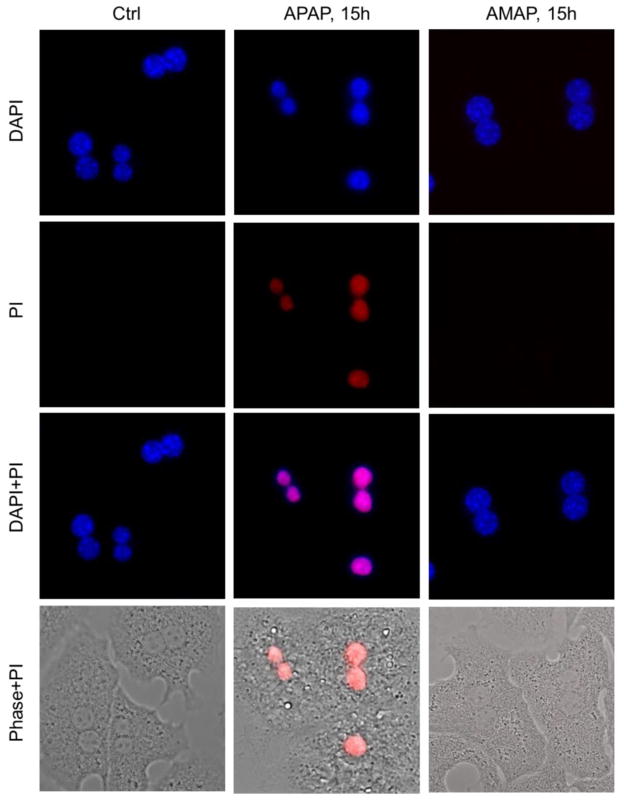
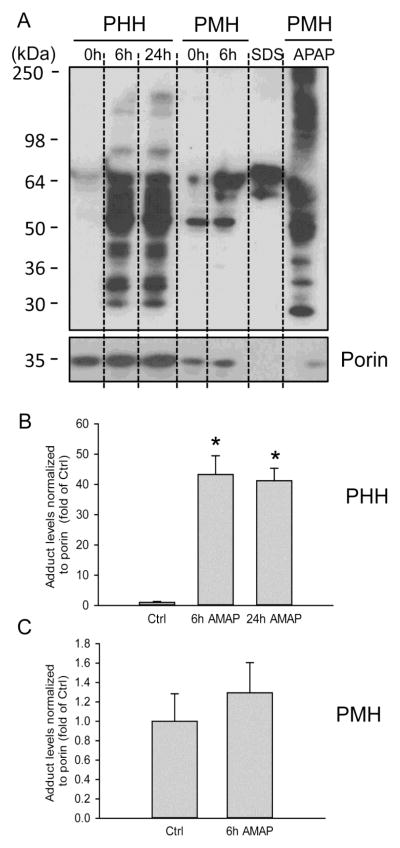
3'-Hydroxyacetanilide orN-acetyl-meta-aminophenol (AMAP) is generally regarded as a non-hepatotoxic analog of acetaminophen (APAP). Previous studies demonstrated the absence of toxicity after AMAP in mice, hamsters, primary mouse hepatocytes and several cell lines. In contrast, experiments with liver slices suggested that it may be toxic to human hepatocytes; however, the mechanism of toxicity is unclear. To explore this,we treated primary human hepatocytes (PHH) with AMAP or APAP for up to 48 h and measured several parameters to assess metabolism and injury. Although less toxic than APAP, AMAP dose-dependently triggered cell death in PHH as indicated by alanine aminotransferase (ALT) release and propidium iodide (PI) staining. Similar to APAP, AMAP also significantly depleted glutathione (GSH) in PHH and caused mitochondrial damage as indicated by glutamate dehydrogenase (GDH) release and the JC-1 assay. However, unlike APAP, AMAP treatment did not cause relevant c-jun-N-terminal kinase (JNK) activation in the cytosol or phospho-JNK translocation to mitochondria. To compare, AMAP toxicity was assessed in primary mouse hepatocytes (PMH). No cytotoxicity was observed as indicated by the lack of lactate dehydrogenase release and no PI staining. Furthermore, there was no GSH depletion or mitochondrial dysfunction after AMAP treatment in PMH. Immunoblotting for arylated proteins suggested that AMAP treatment caused extensive mitochondrial protein adduct formation in PHH but not in PMH. In conclusion, AMAP is hepatotoxic in PHH and the mechanism involves the formation of mitochondrial protein adducts and mitochondrial dysfunction.
Keywords: 3′-Hydroxyacetanilide (AMAP); Acetaminophen; Hepatotoxicity; Mitochondrial dysfunction; Protein adducts.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Bajt ML, Lawson JA, Vonderfecht SL, Gujral JS, Jaeschke H. Protection against Fas receptor-mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: evidence for a postmitochondrial processing of caspase-8. Toxicol Sci. 2000;58:109–17. - PubMed
-
- Bajt ML, Cover C, Lemasters JJ, Jaeschke H. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol Sci. 2006;94:217–25. - PubMed
-
- Bajt ML, Farhood A, Lemasters JJ, Jaeschke H. Mitochondrial bax translocation accelerates DNA fragmentation and cell necrosis in a murine model of acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2008;324:8–14. - PubMed
-
- Bajt ML, Knight TR, Lemasters JJ, Jaeschke H. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: protection by N-acetyl cysteine. Toxicol Sci. 2004;80:343–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
