

UBA1: At the Crossroads of Ubiquitin Homeostasis and Neurodegeneration
- PMID: 26432019
- PMCID: PMC4596250
- DOI: 10.1016/j.molmed.2015.08.003
UBA1: At the Crossroads of Ubiquitin Homeostasis and Neurodegeneration
Abstract
Neurodegenerative diseases are a leading cause of disability and early death. A common feature of these conditions is disruption of protein homeostasis. Ubiquitin-like modifier activating enzyme 1 (UBA1), the E1 ubiquitin-activating enzyme, sits at the apex of the ubiquitin cascade and represents an important regulator of cellular protein homeostasis. Critical contributions of UBA1-dependent pathways to the regulation of homeostasis and degeneration in the nervous system are emerging, including specific disruption of UBA1 in spinal muscular atrophy (SMA) and Huntington's disease (HD). In this review we discuss recent findings that put UBA1 at the centre of cellular homeostasis and neurodegeneration, highlighting the potential for UBA1 to act as a promising therapeutic target for a range of neurodegenerative diseases.
Copyright © 2015 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures
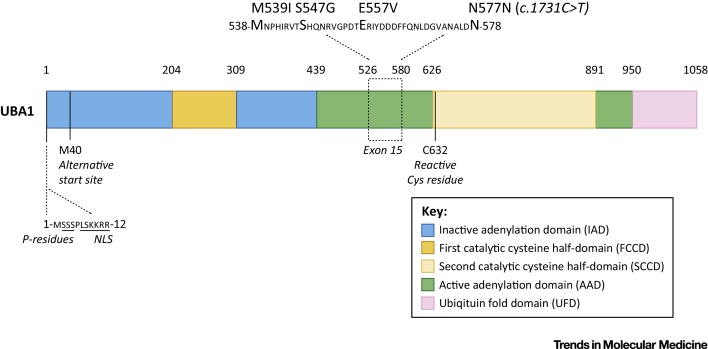
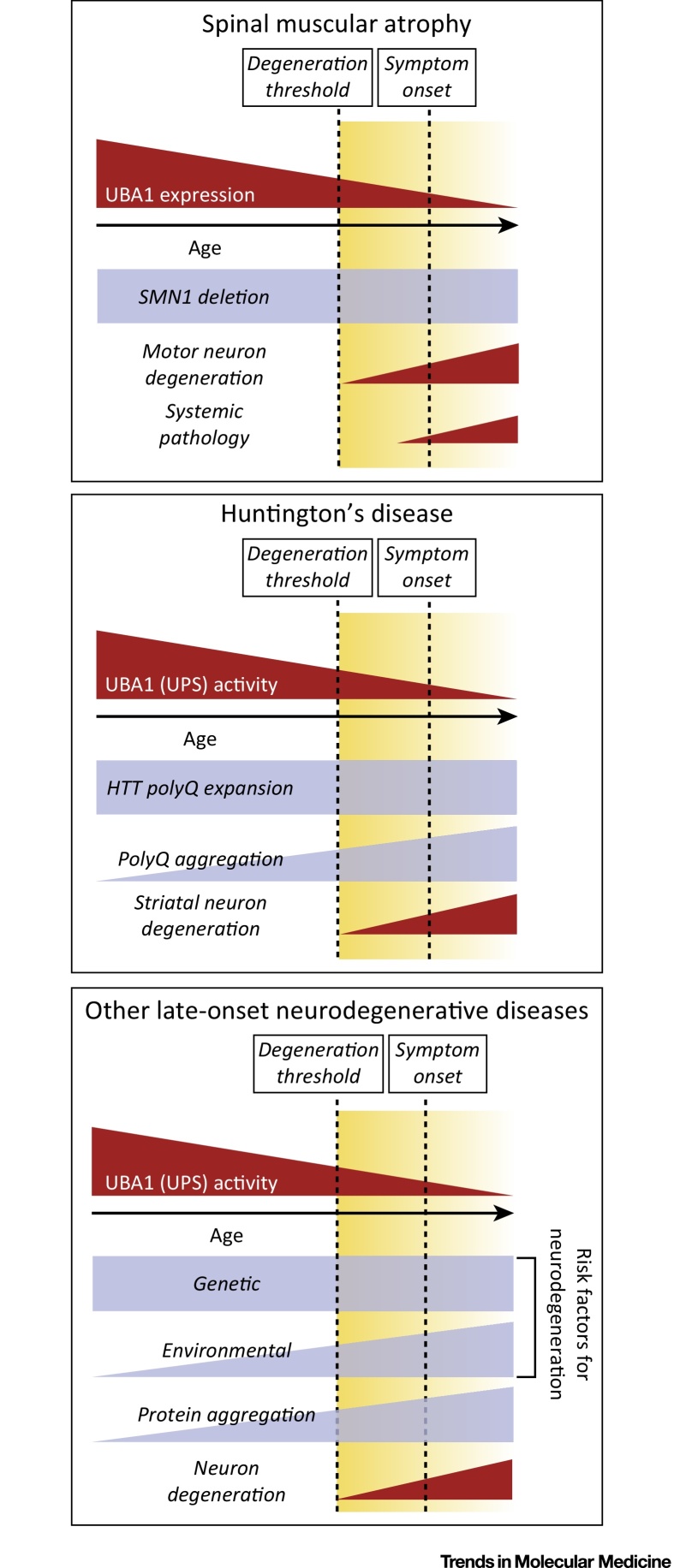
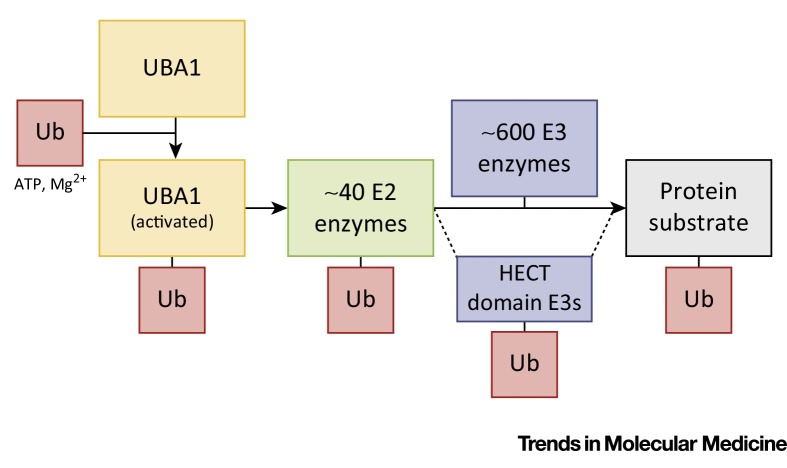
References
-
- Schapira A.H. Slowing of neurodegeneration in Parkinson's disease and Huntington's disease: future therapeutic perspectives. Lancet. 2014;384:545–555. - PubMed
-
- van den Berg L.H. Therapy of amyotrophic lateral sclerosis remains a challenge. Lancet Neurol. 2014;13:1062–1063. - PubMed
-
- Mitsui J., Tsuji S. Genomic aspects of sporadic neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2014;452:221–225. - PubMed
-
- Rubinsztein D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
