

Reactive oxygen species and mitochondria: A nexus of cellular homeostasis
- PMID: 26432659
- PMCID: PMC4596921
- DOI: 10.1016/j.redox.2015.09.005
Reactive oxygen species and mitochondria: A nexus of cellular homeostasis
Abstract
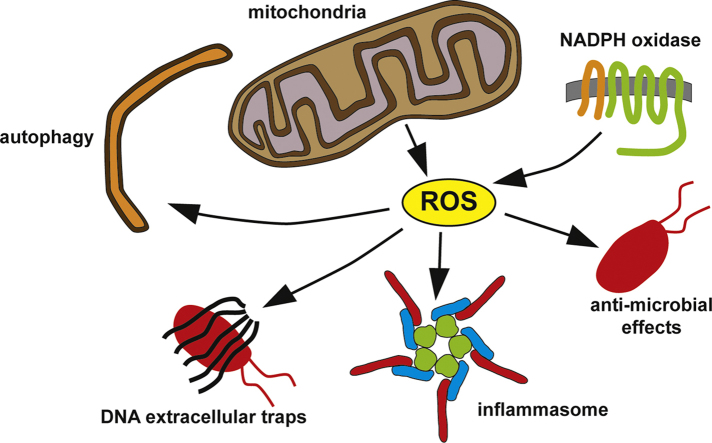
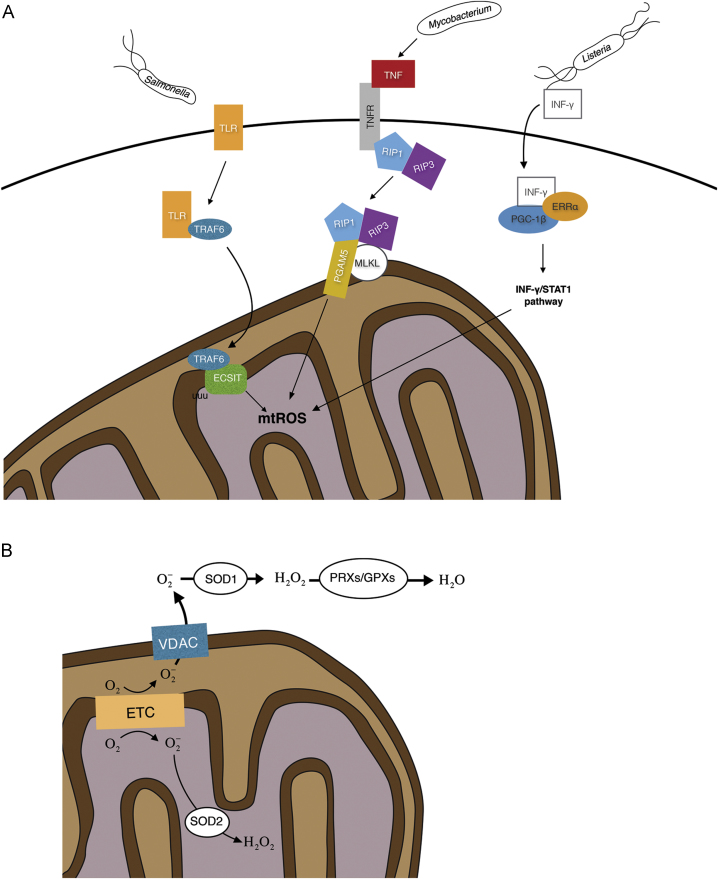
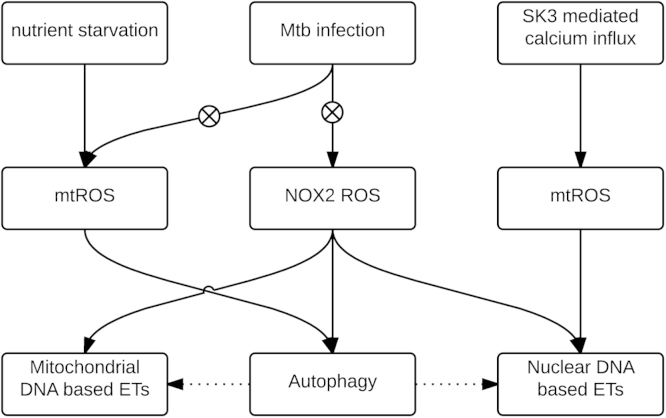
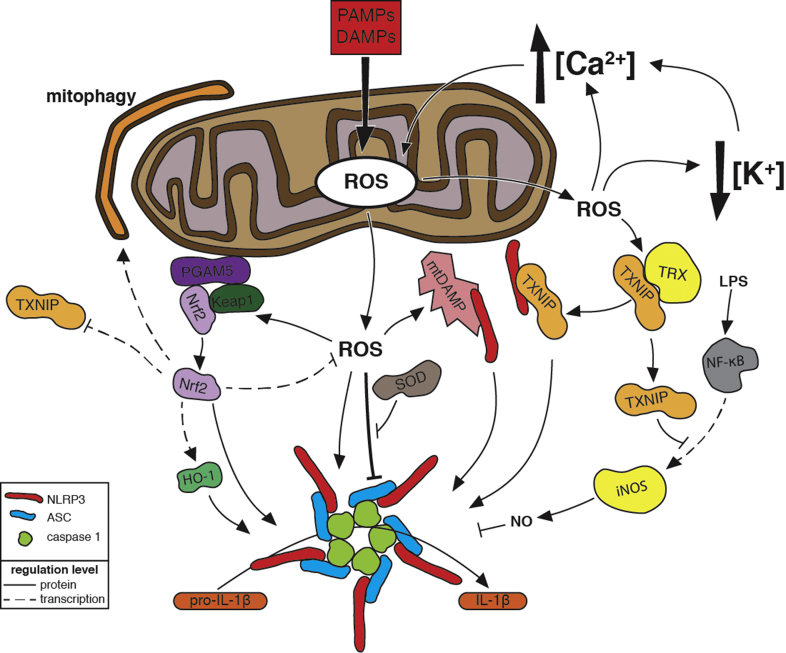
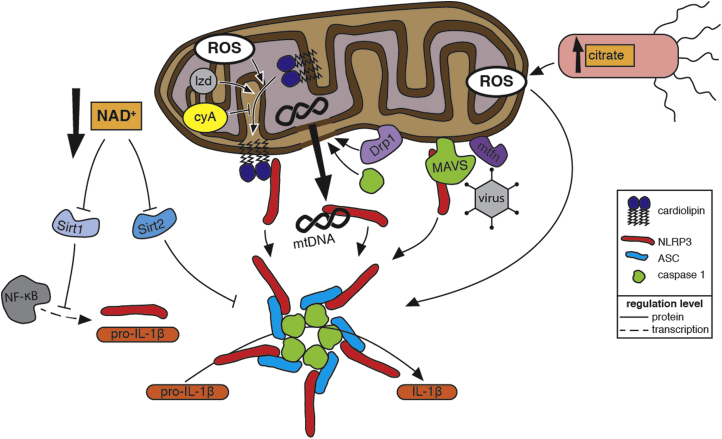
Reactive oxygen species (ROS) are integral components of multiple cellular pathways even though excessive or inappropriately localized ROS damage cells. ROS function as anti-microbial effector molecules and as signaling molecules that regulate such processes as NF-kB transcriptional activity, the production of DNA-based neutrophil extracellular traps (NETs), and autophagy. The main sources of cellular ROS are mitochondria and NADPH oxidases (NOXs). In contrast to NOX-generated ROS, ROS produced in the mitochondria (mtROS) were initially considered to be unwanted by-products of oxidative metabolism. Increasing evidence indicates that mtROS have been incorporated into signaling pathways including those regulating immune responses and autophagy. As metabolic hubs, mitochondria facilitate crosstalk between the metabolic state of the cell with these pathways. Mitochondria and ROS are thus a nexus of multiple pathways that determine the response of cells to disruptions in cellular homeostasis such as infection, sterile damage, and metabolic imbalance. In this review, we discuss the roles of mitochondria in the generation of ROS-derived anti-microbial effectors, the interplay of mitochondria and ROS with autophagy and the formation of DNA extracellular traps, and activation of the NLRP3 inflammasome by ROS and mitochondria.
Keywords: Autophagy; Immunity; Inflammasome; Mitochondria; Neutrophil extracellular traps; Reactive oxygen species.
Copyright © 2015. Published by Elsevier B.V.
Figures
References
-
- Lam G.Y., Huang J., Brumell J.H. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin Immunopathol. 2010;32:415–430. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
