

Signaling pathway and dysregulation of PD1 and its ligands in lymphoid malignancies
- PMID: 26432723
- PMCID: PMC4733614
- DOI: 10.1016/j.bbcan.2015.09.002
Signaling pathway and dysregulation of PD1 and its ligands in lymphoid malignancies
Abstract
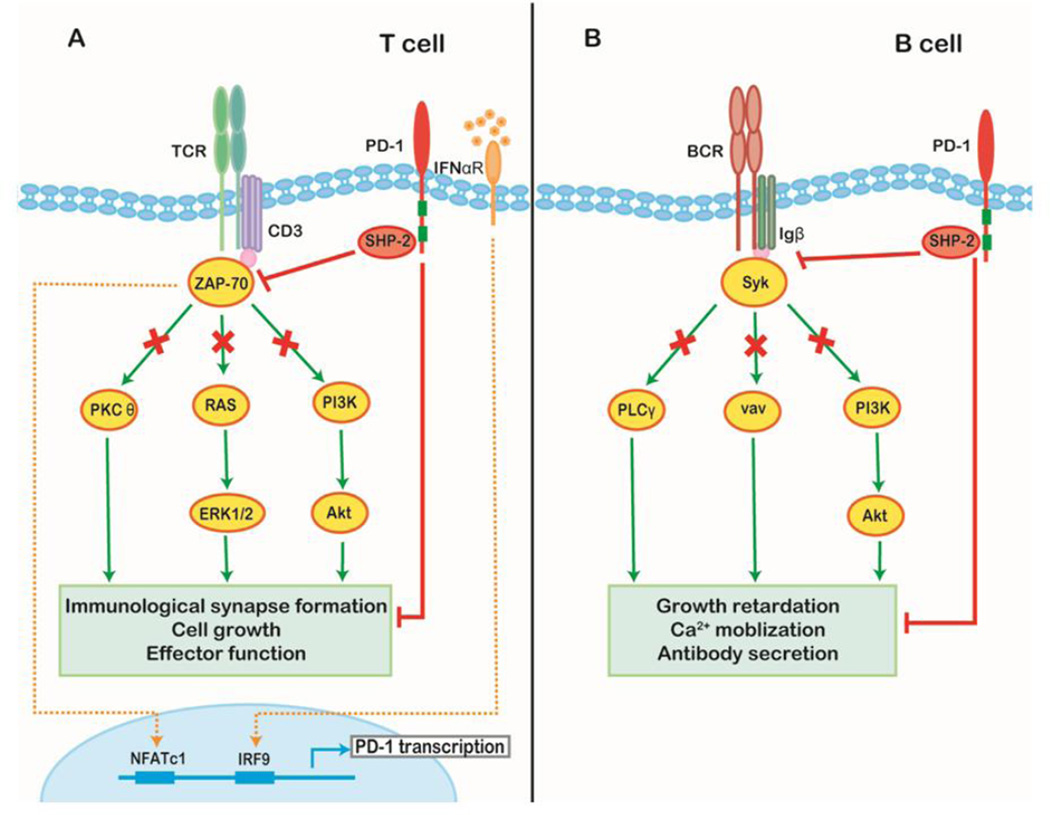
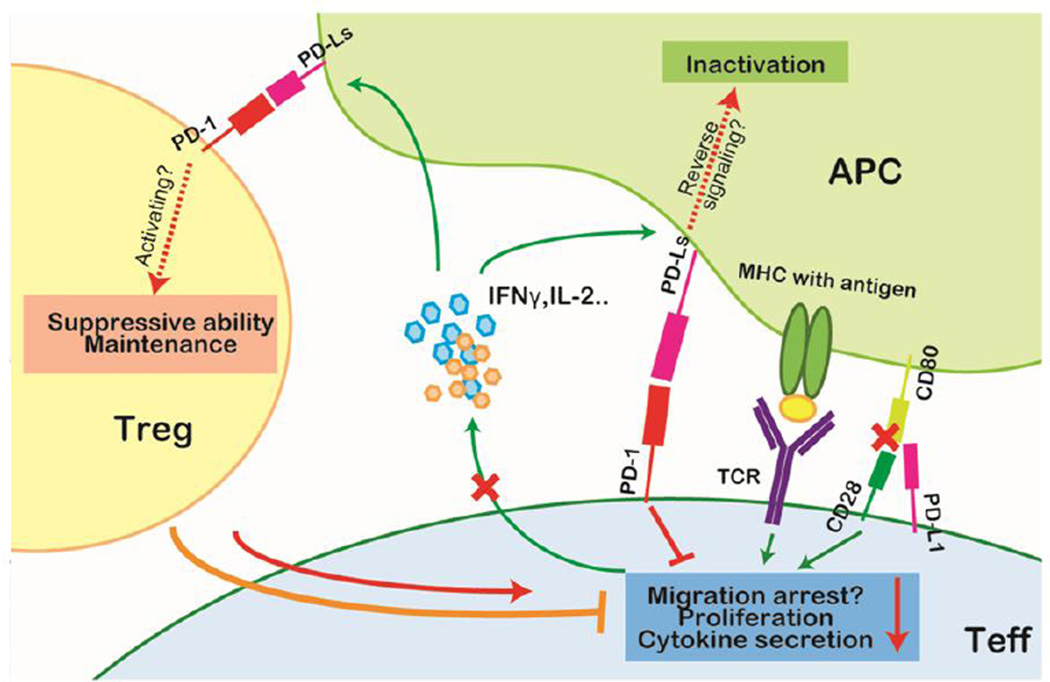
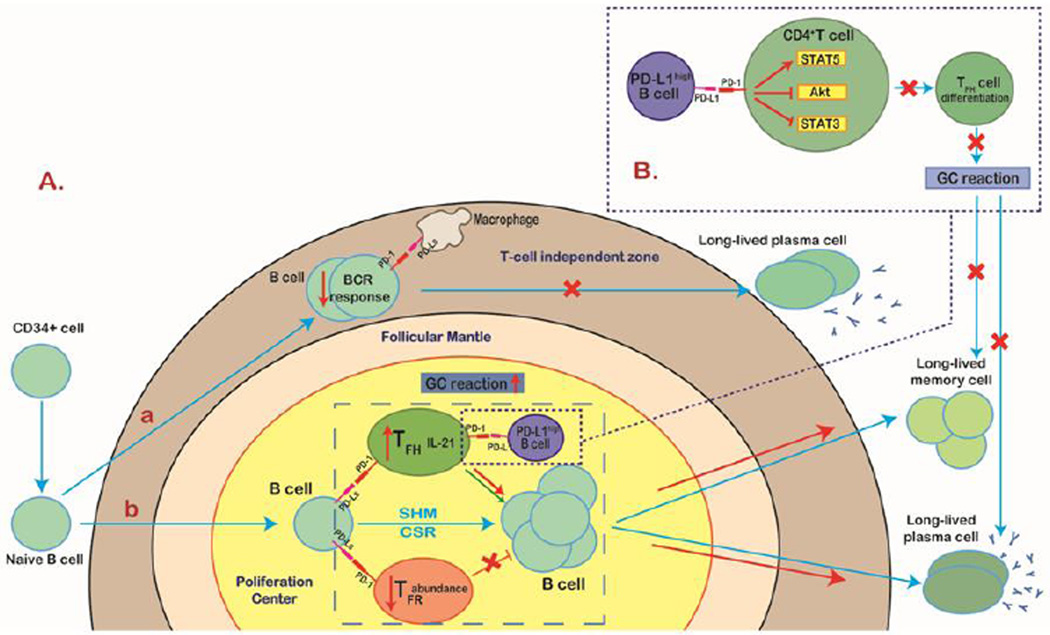
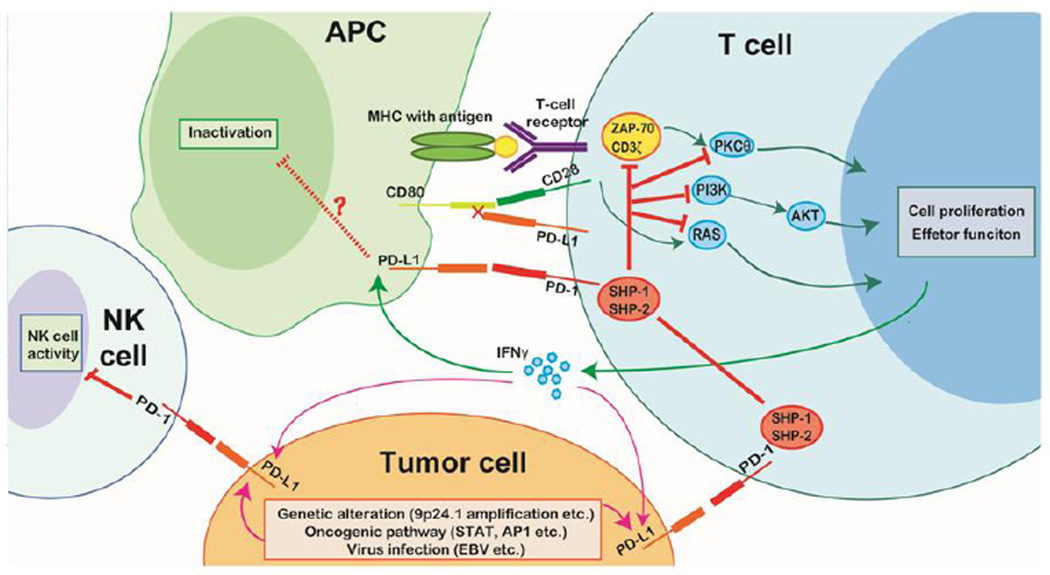
Tumor cells evade immune destruction, at least partially, by upregulating inhibitory signals to limit effector T cell activation. Programmed death 1 (PD-1) is one of the most critical co-inhibitory molecules limiting the T-cell antitumor response. PD-1 and its ligands, PD-L1 and PD-L2, are overexpressed by various types of tumors as well as reactive cells in the tumor microenvironment. A growing body of evidence has shown the clinical efficiency and minimal toxicity of PD-1 pathway inhibitors in patients with solid tumors, but the role of these inhibitors in lymphoid malignancies is much less well studied. In this review, we analyze the pathologic role of the PD-1 pathway in most common lymphoid malignancies and we organize the clinical data from clinical trials of PD-1 pathway inhibitors. Several anti-PD-1 regimens have shown encouraging therapeutic effects in patients with relapsed or refractory Hodgkin lymphoma, follicular lymphoma, and diffuse large B-cell lymphoma. Additional progress is needed to foster an improved understanding of the role of anti-PD-1 therapy in reconstituting antitumor immunity in patients with lymphoid malignancies. Upcoming trials will explore the clinical efficiency of combining PD-1 pathway inhibitors and various agents with diverse mechanisms of action and create more therapeutic possibilities for afflicted patients.
Keywords: Lymphoid malignancies; Nivolumab; PD-1; PD-L1; PD-L2; Pembrolizumab; Pidilizumab.
Copyright © 2015 Elsevier B.V. All rights reserved.
Figures
References
-
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. - PubMed
-
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
