

Population genomic structure and adaptation in the zoonotic malaria parasite Plasmodium knowlesi
- PMID: 26438871
- PMCID: PMC4620865
- DOI: 10.1073/pnas.1509534112
Population genomic structure and adaptation in the zoonotic malaria parasite Plasmodium knowlesi
Abstract
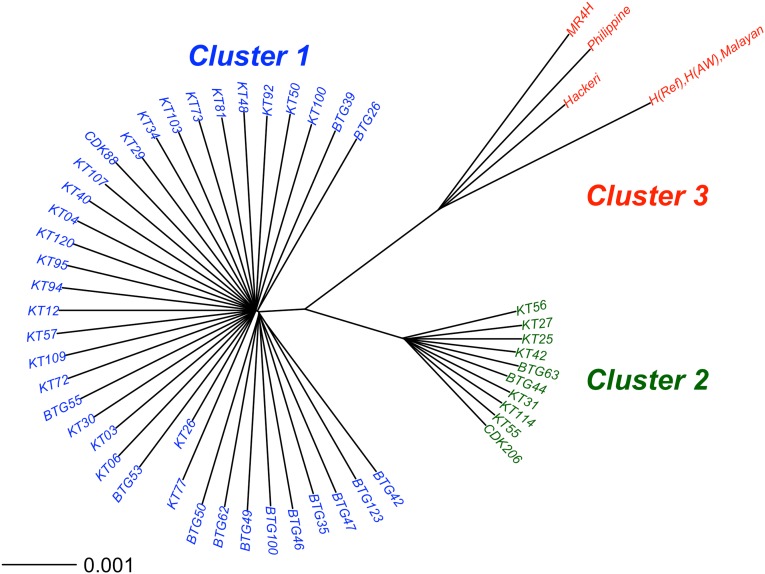
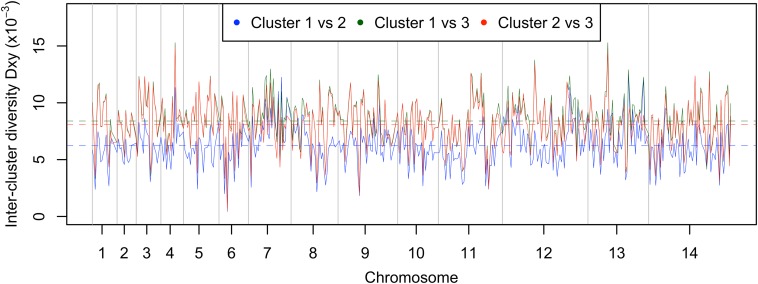
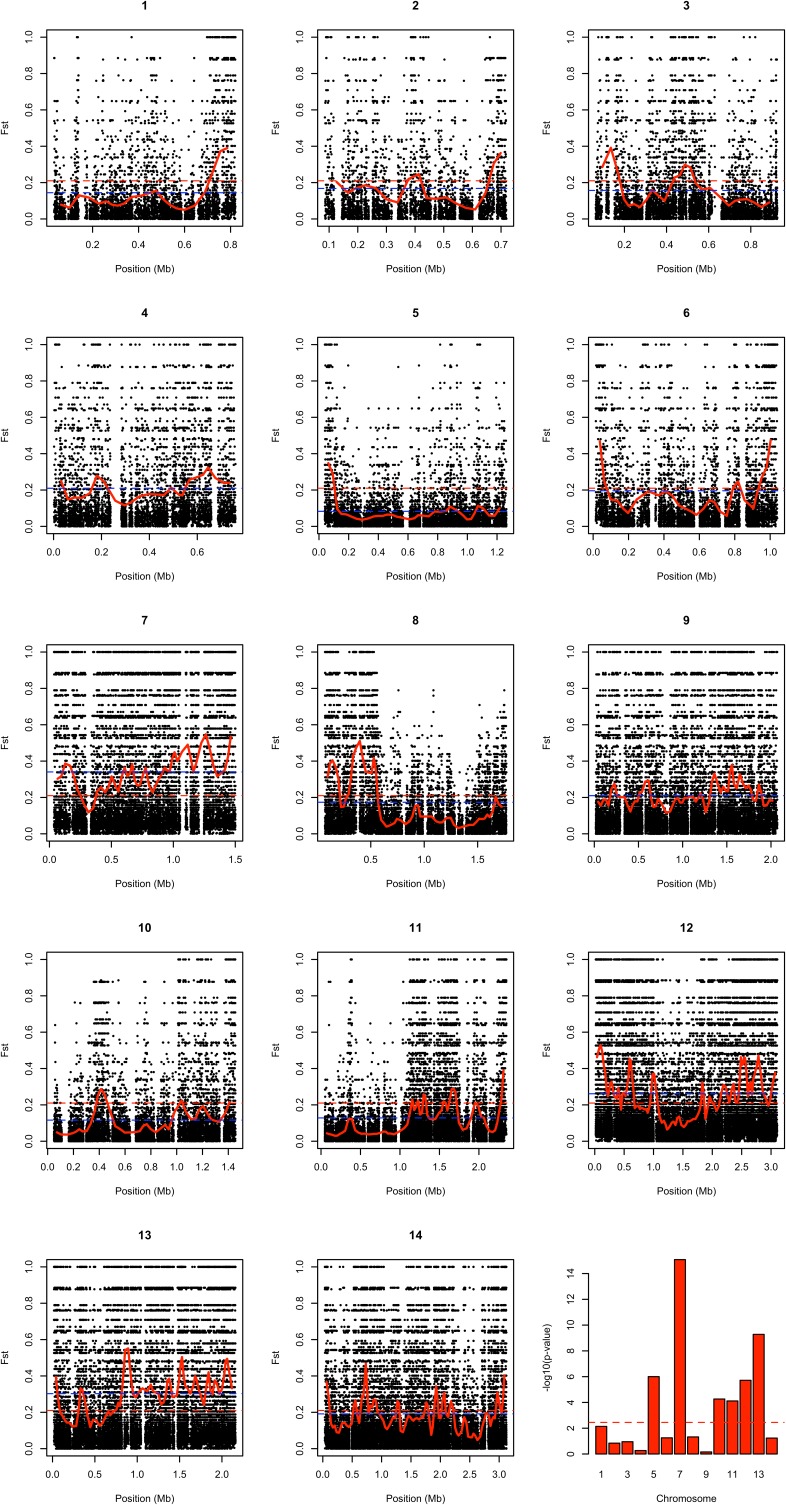
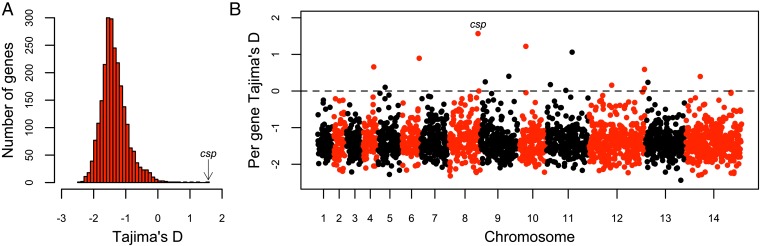
Malaria cases caused by the zoonotic parasite Plasmodium knowlesi are being increasingly reported throughout Southeast Asia and in travelers returning from the region. To test for evidence of signatures of selection or unusual population structure in this parasite, we surveyed genome sequence diversity in 48 clinical isolates recently sampled from Malaysian Borneo and in five lines maintained in laboratory rhesus macaques after isolation in the 1960s from Peninsular Malaysia and the Philippines. Overall genomewide nucleotide diversity (π = 6.03 × 10(-3)) was much higher than has been seen in worldwide samples of either of the major endemic malaria parasite species Plasmodium falciparum and Plasmodium vivax. A remarkable substructure is revealed within P. knowlesi, consisting of two major sympatric clusters of the clinical isolates and a third cluster comprising the laboratory isolates. There was deep differentiation between the two clusters of clinical isolates [mean genomewide fixation index (FST) = 0.21, with 9,293 SNPs having fixed differences of FST = 1.0]. This differentiation showed marked heterogeneity across the genome, with mean FST values of different chromosomes ranging from 0.08 to 0.34 and with further significant variation across regions within several chromosomes. Analysis of the largest cluster (cluster 1, 38 isolates) indicated long-term population growth, with negatively skewed allele frequency distributions (genomewide average Tajima's D = -1.35). Against this background there was evidence of balancing selection on particular genes, including the circumsporozoite protein (csp) gene, which had the top Tajima's D value (1.57), and scans of haplotype homozygosity implicate several genomic regions as being under recent positive selection.
Keywords: Plasmodium diversity; adaptation; population genomics; reproductive isolation; zoonosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- Garnham P. Malaria Parasites and Other Haemosporidia. Blackwell Scientific Publications Ltd.; Oxford, UK: 1966.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
