

Detecting Horizontal Gene Transfer between Closely Related Taxa
- PMID: 26439115
- PMCID: PMC4595140
- DOI: 10.1371/journal.pcbi.1004408
Detecting Horizontal Gene Transfer between Closely Related Taxa
Abstract
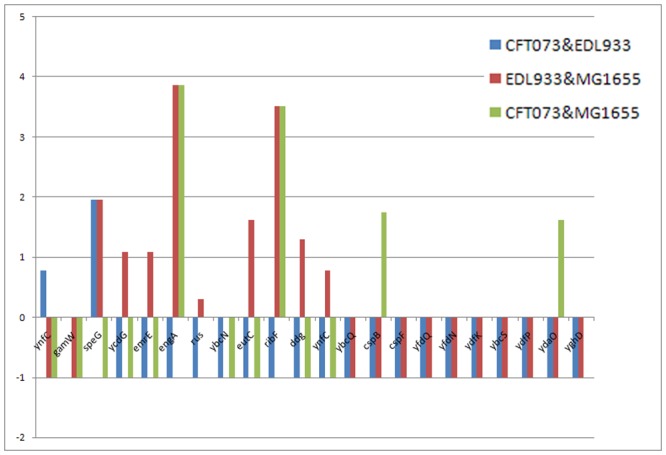
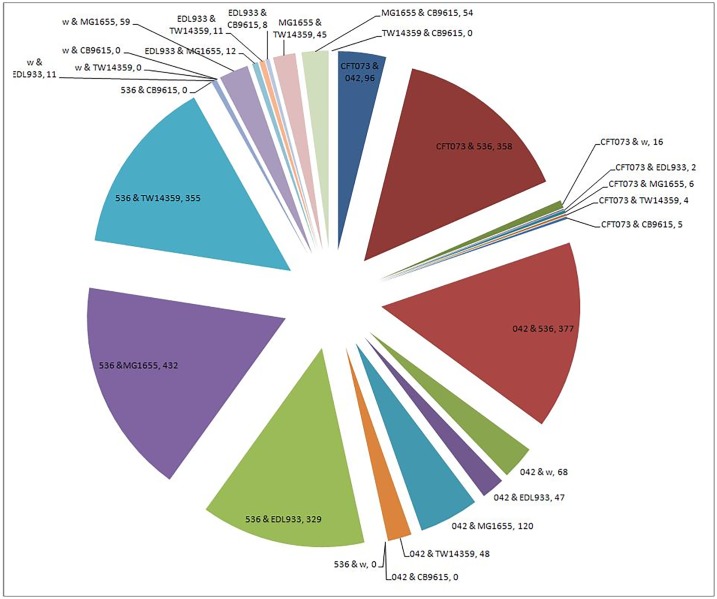
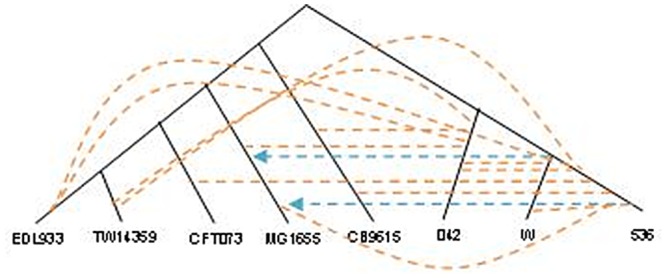
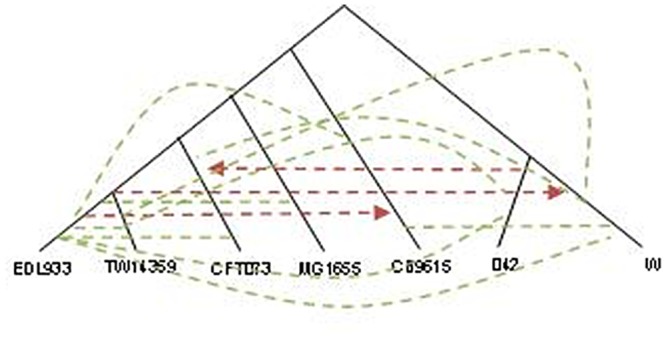
Horizontal gene transfer (HGT), the transfer of genetic material between organisms, is crucial for genetic innovation and the evolution of genome architecture. Existing HGT detection algorithms rely on a strong phylogenetic signal distinguishing the transferred sequence from ancestral (vertically derived) genes in its recipient genome. Detecting HGT between closely related species or strains is challenging, as the phylogenetic signal is usually weak and the nucleotide composition is normally nearly identical. Nevertheless, there is a great importance in detecting HGT between congeneric species or strains, especially in clinical microbiology, where understanding the emergence of new virulent and drug-resistant strains is crucial, and often time-sensitive. We developed a novel, self-contained technique named Near HGT, based on the synteny index, to measure the divergence of a gene from its native genomic environment and used it to identify candidate HGT events between closely related strains. The method confirms candidate transferred genes based on the constant relative mutability (CRM). Using CRM, the algorithm assigns a confidence score based on "unusual" sequence divergence. A gene exhibiting exceptional deviations according to both synteny and mutability criteria, is considered a validated HGT product. We first employed the technique to a set of three E. coli strains and detected several highly probable horizontally acquired genes. We then compared the method to existing HGT detection tools using a larger strain data set. When combined with additional approaches our new algorithm provides richer picture and brings us closer to the goal of detecting all newly acquired genes in a particular strain.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
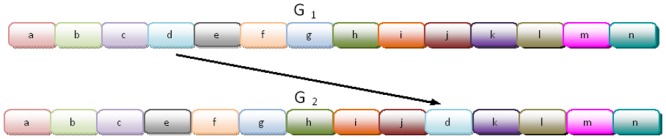
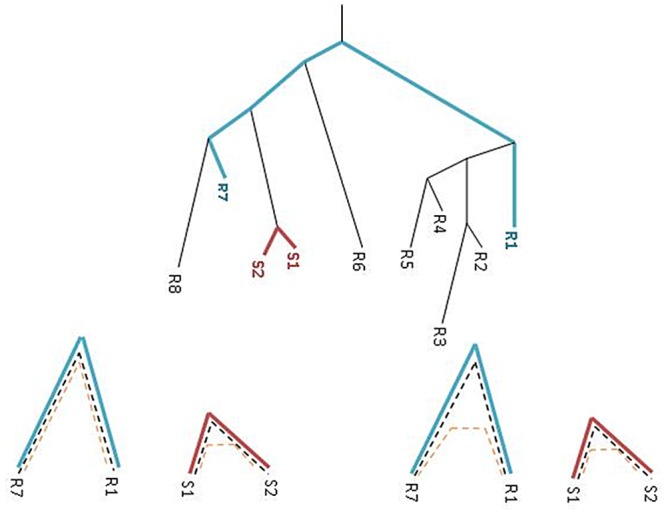
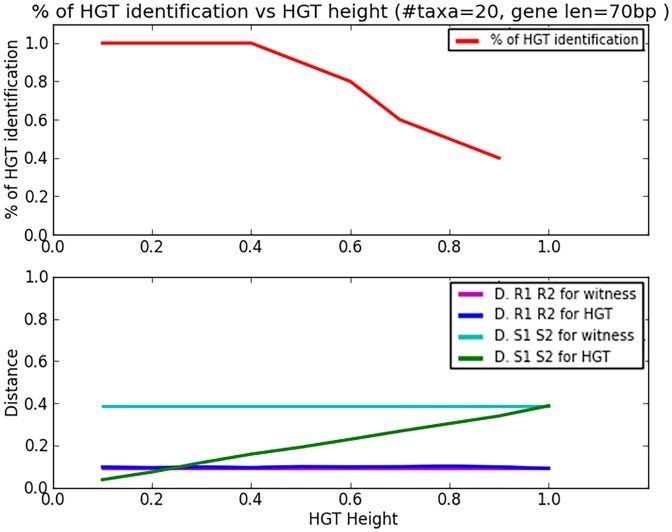
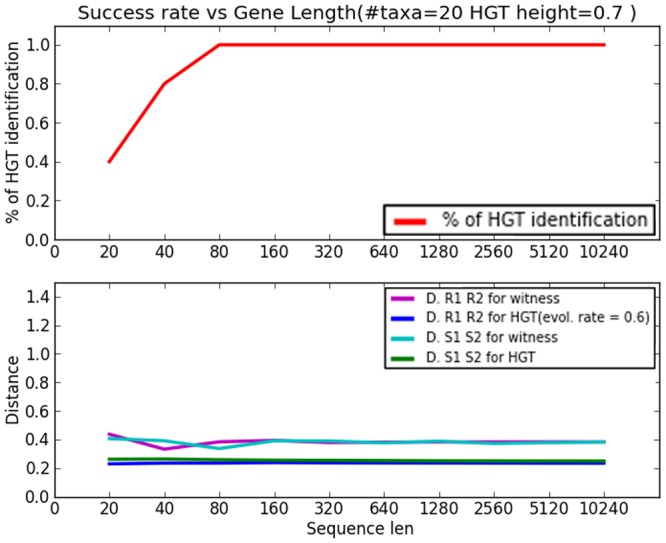
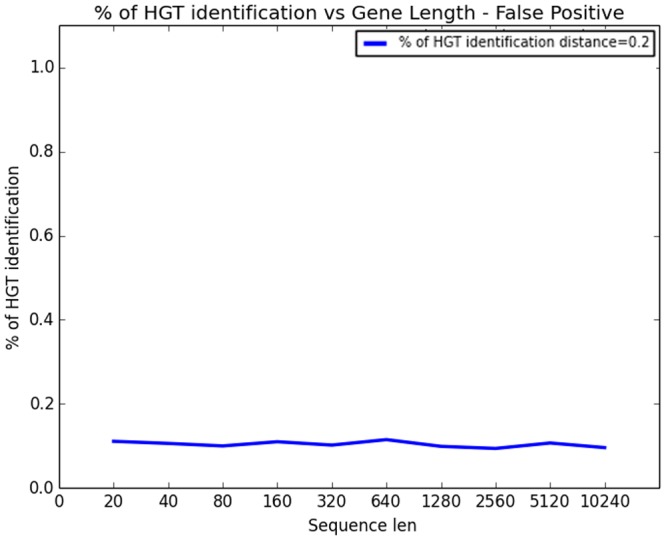
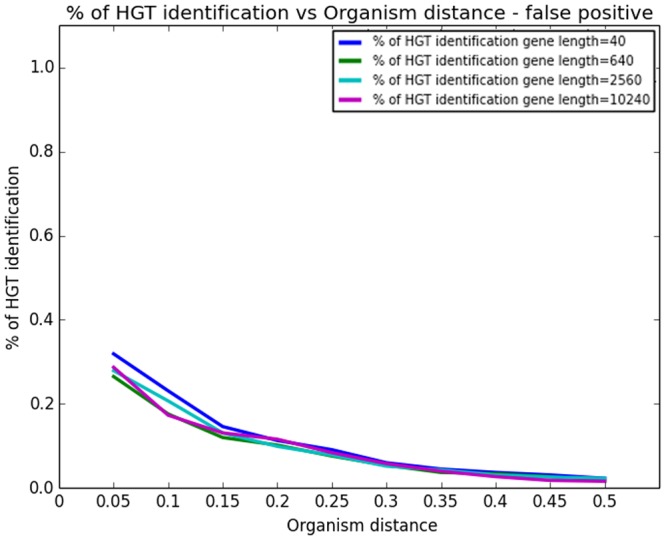
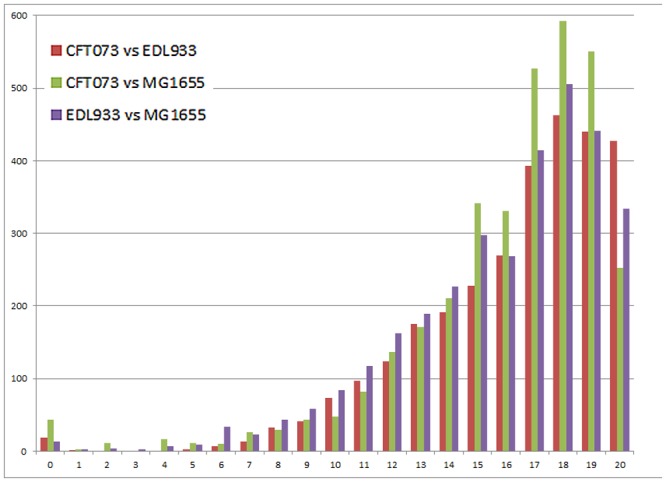
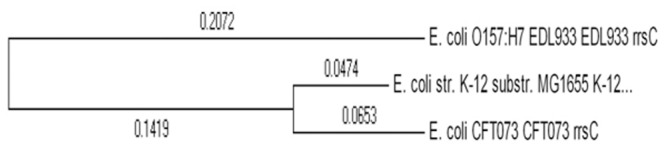
References
-
- Koonin E. V. and Galperin M. Y., Sequence—Evolution—Function. Computational Approaches in Comparative Genomics. Springer, 2002. - PubMed
-
- Gogarten J. and Townsend J., “Horizontal gene transfer, genome innovation and evolution,” Nat Rev Microbiol., vol. 3, no. 9, pp. 679–87, 2005. - PubMed
-
- Doolittle W. F., “Phylogenetic classification and the universal tree,” Science, vol. 284, no. 5423, pp. 2124–9, 1999. - PubMed
-
- Nakamura Y., Itoh T., Matsuda H., and Gojobori T., “Biased biological functions of horizontally transferred genes in prokaryotic genomes,” Nat Genet, vol. 36, no. 7, pp. 760–6, 2004. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
