

Genetic variation and the de novo assembly of human genomes
- PMID: 26442640
- PMCID: PMC4745987
- DOI: 10.1038/nrg3933
Genetic variation and the de novo assembly of human genomes
Abstract
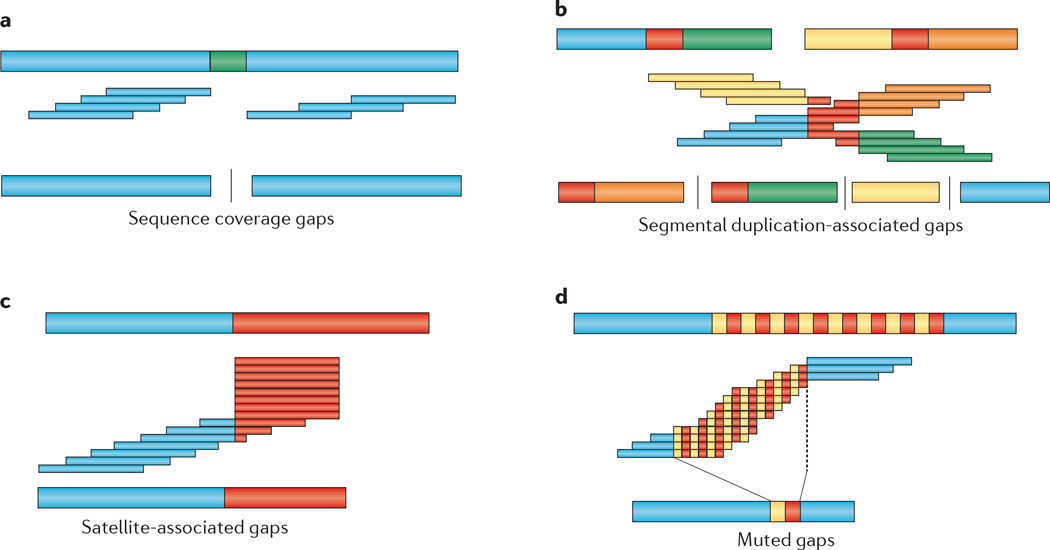
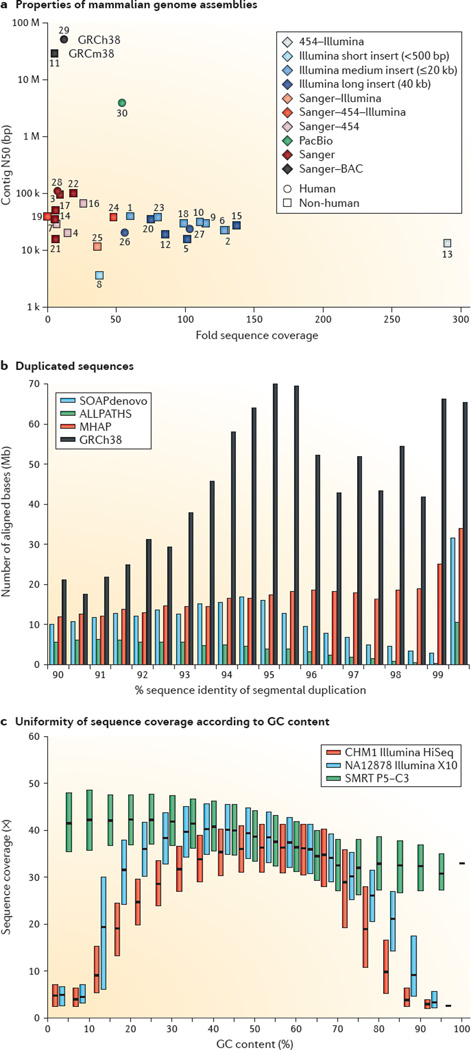
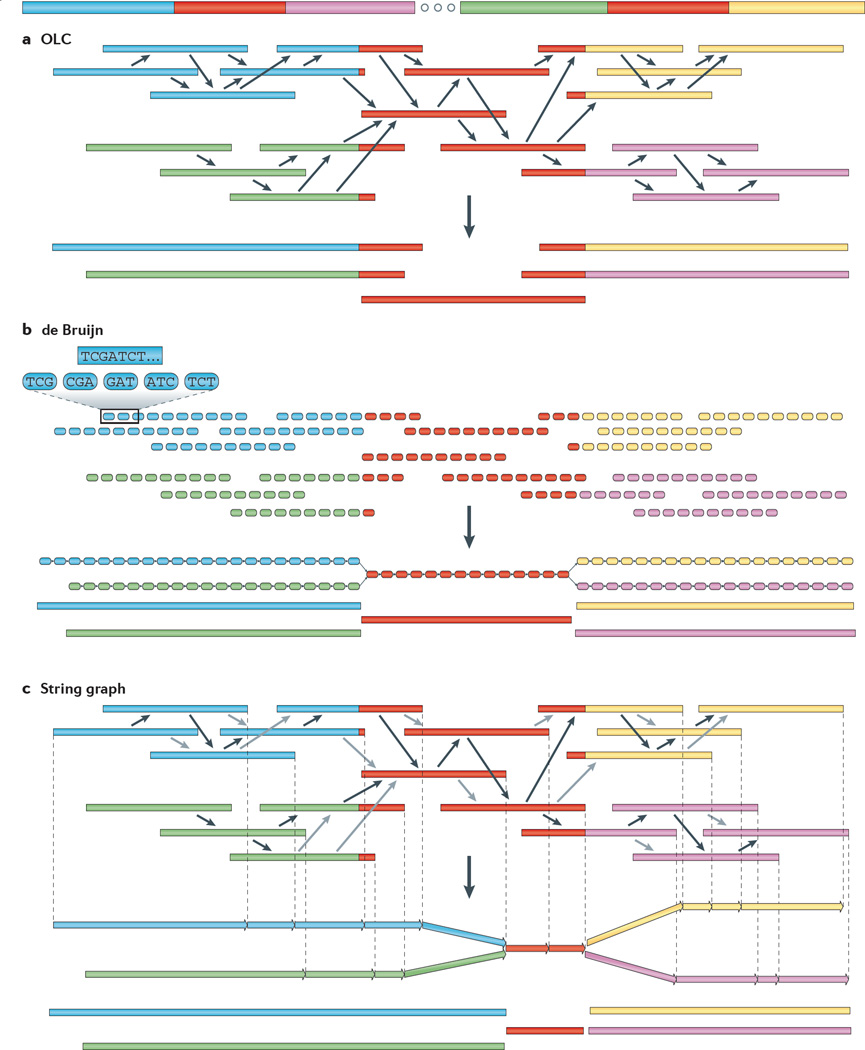
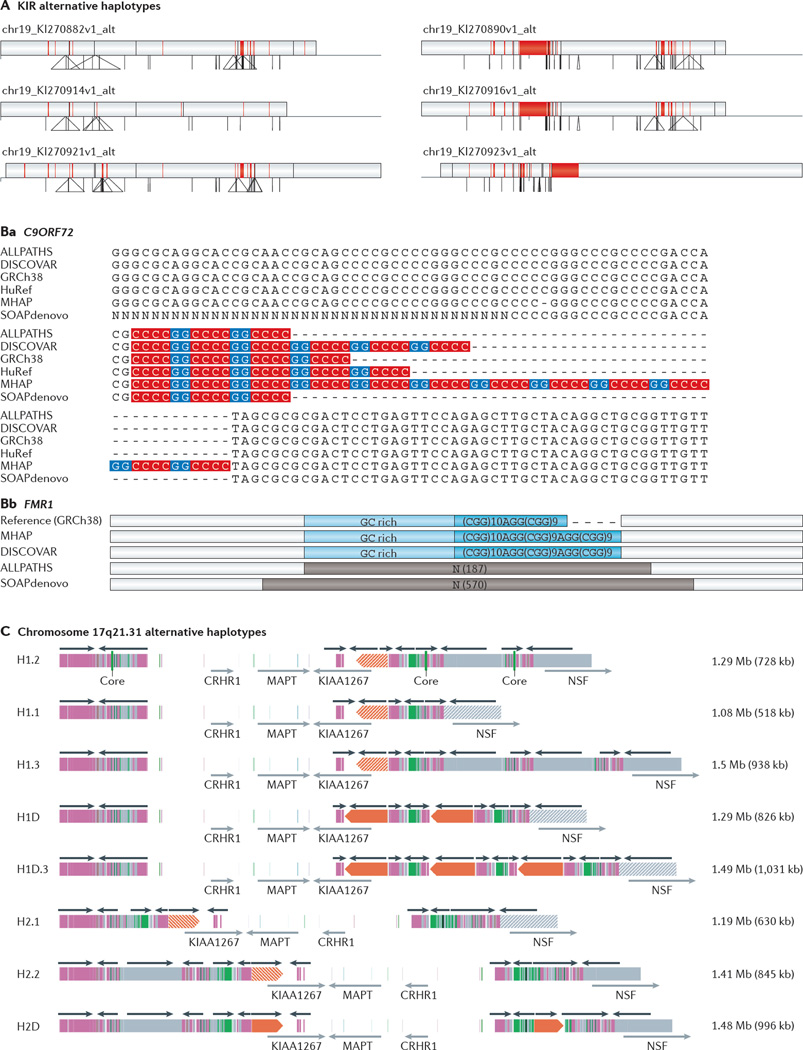
The discovery of genetic variation and the assembly of genome sequences are both inextricably linked to advances in DNA-sequencing technology. Short-read massively parallel sequencing has revolutionized our ability to discover genetic variation but is insufficient to generate high-quality genome assemblies or resolve most structural variation. Full resolution of variation is only guaranteed by complete de novo assembly of a genome. Here, we review approaches to genome assembly, the nature of gaps or missing sequences, and biases in the assembly process. We describe the challenges of generating a complete de novo genome assembly using current technologies and the impact that being able to perfectly sequence the genome would have on understanding human disease and evolution. Finally, we summarize recent technological advances that improve both contiguity and accuracy and emphasize the importance of complete de novo assembly as opposed to read mapping as the primary means to understanding the full range of human genetic variation.
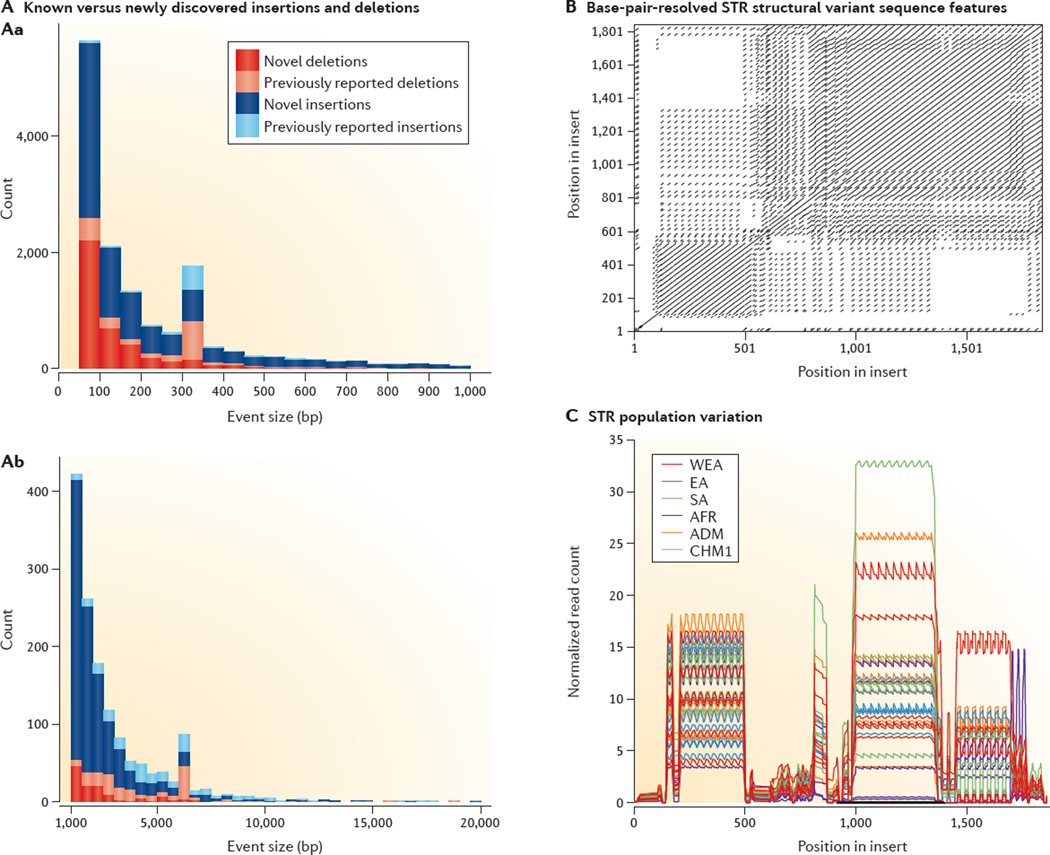
Figures
References
-
-
Chaisson MJP, et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature. 2015;517:608–611. Long-read sequencing paired with local assembly reveals structural variation and closes or extends ~50% of the gaps in the reference human genome.
-
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
