

Genetics of Hearing Loss: Syndromic
- PMID: 26443487
- PMCID: PMC4641804
- DOI: 10.1016/j.otc.2015.07.007
Genetics of Hearing Loss: Syndromic
Abstract
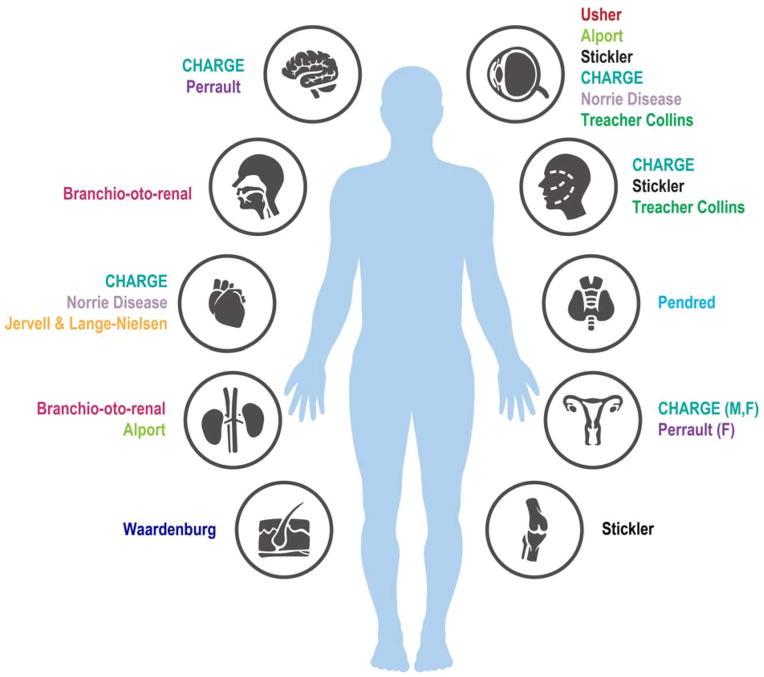
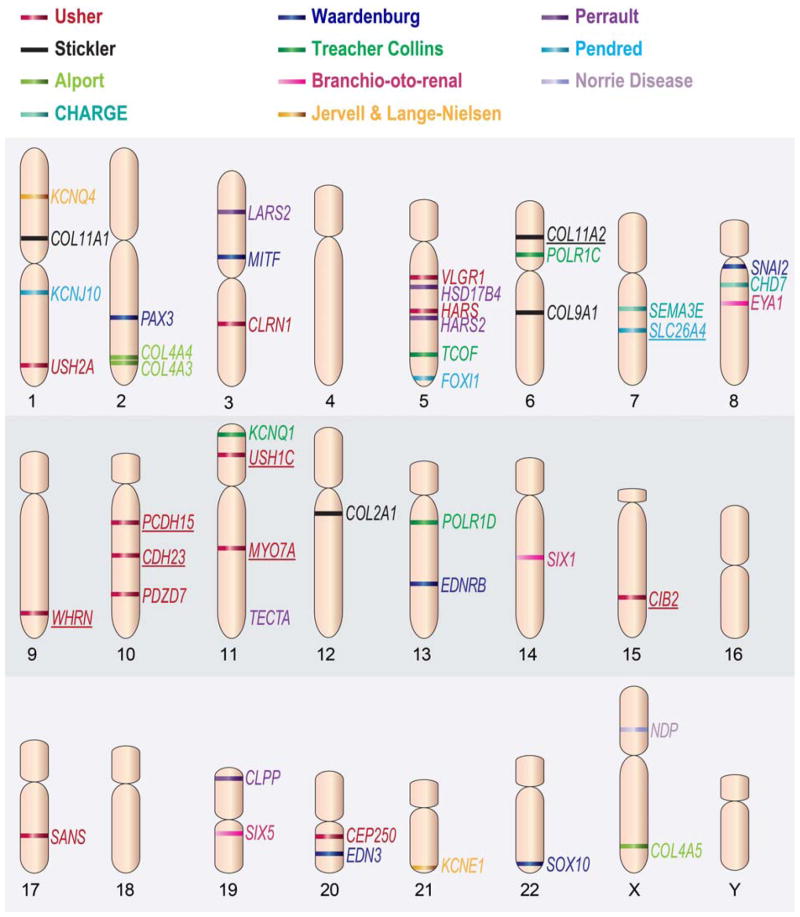
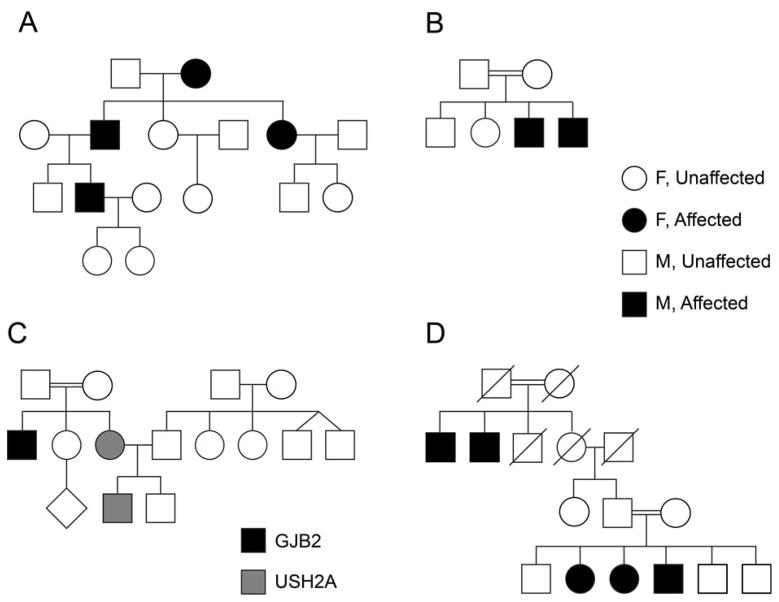
Hearing loss (HL) is one of the most common birth defects in developed countries and is a diverse pathologic condition with different classifications. One of these is based on the association with other clinical features, defined as syndromic hearing loss (SHL). Determining the cause of the HL in these patients is extremely beneficial as it enables a personalized approach to caring for the individual. Early screening can further aid in optimal rehabilitation for a child's development and growth. The advancement of high-throughput sequencing technology is facilitating rapid and low-cost diagnostics for patients with SHL.
Keywords: Deafness; Genetics; Genome; Hearing loss; Sequencing.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
References
-
- Quaranta N, Coppola F, Casulli M, et al. Epidemiology of age related hearing loss: A review. Hearing, Balance and Communication. 2015:1–5.
-
- Morton CC, Nance WE. Newborn hearing screening--a silent revolution. N Engl J Med. 2006;354:2151–2164. - PubMed
-
- Nance WE. The genetics of deafness. Ment Retard Dev Disabil Res Rev. 2003;9:109–119. - PubMed
-
- Dror AA, Avraham KB. Hearing impairment: a panoply of genes and functions. Neuron. 2010;68:293–308. - PubMed
-
- Ross M, Pawlina W. Histology: A Text and Atlas, with Correlated Cell and Molecular Biology. 6. 2010. Paperback.
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
