

Calcineurin mediates homeostatic synaptic plasticity by regulating retinoic acid synthesis
- PMID: 26443861
- PMCID: PMC4620864
- DOI: 10.1073/pnas.1510239112
Calcineurin mediates homeostatic synaptic plasticity by regulating retinoic acid synthesis
Abstract
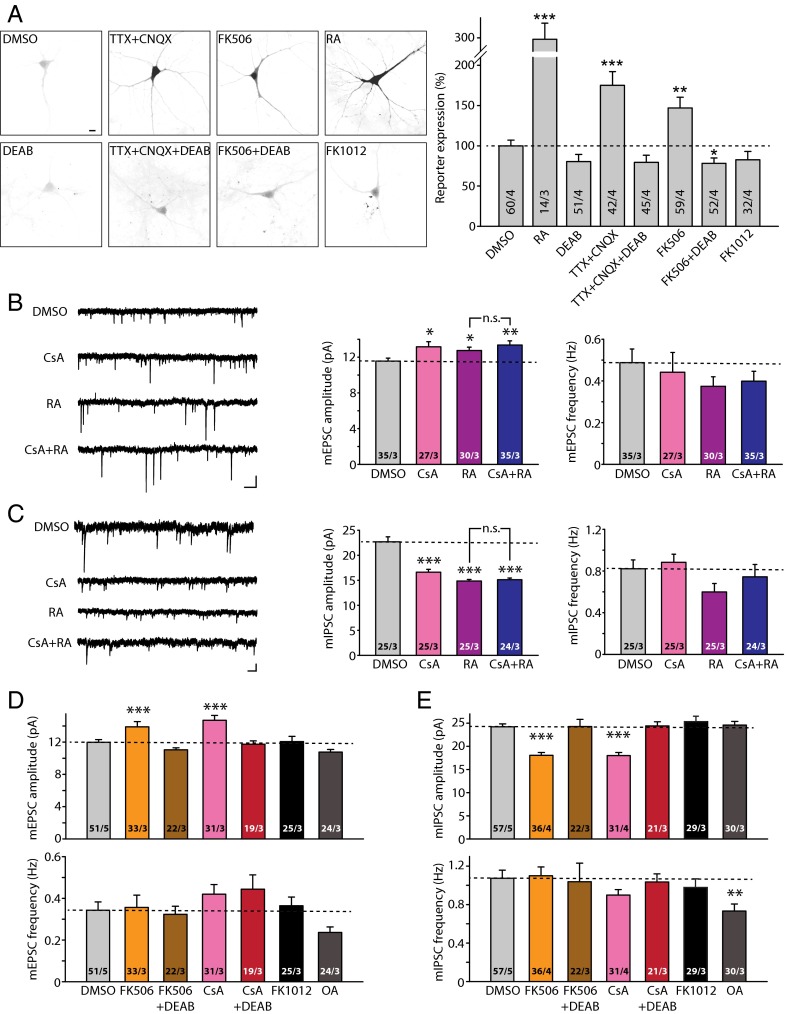
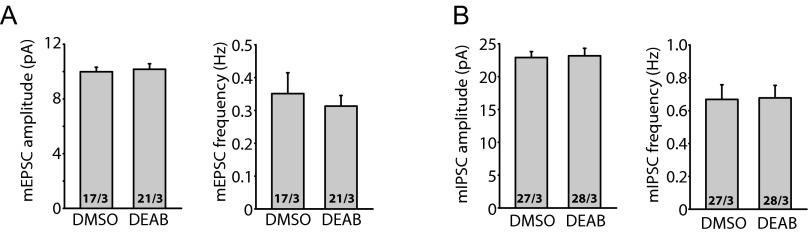
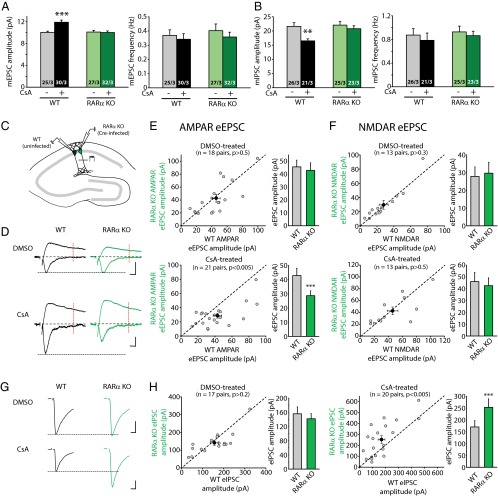
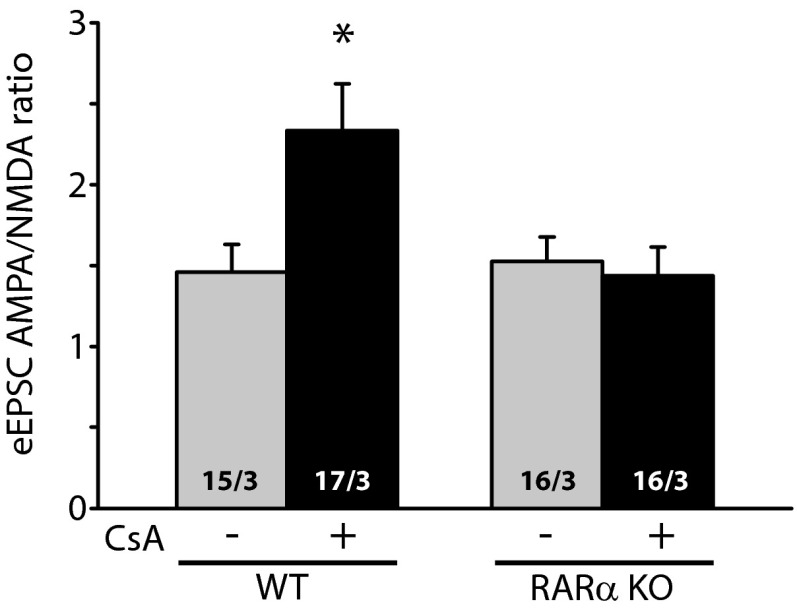
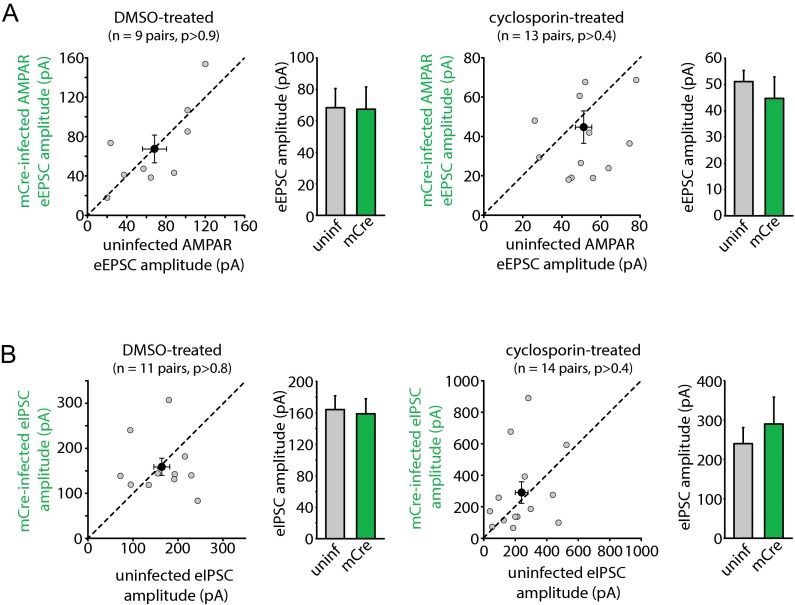
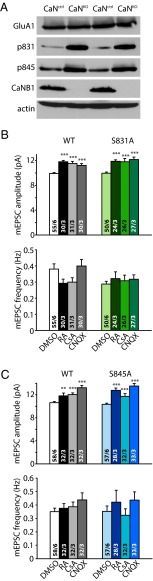
Homeostatic synaptic plasticity is a form of non-Hebbian plasticity that maintains stability of the network and fidelity for information processing in response to prolonged perturbation of network and synaptic activity. Prolonged blockade of synaptic activity decreases resting Ca(2+) levels in neurons, thereby inducing retinoic acid (RA) synthesis and RA-dependent homeostatic synaptic plasticity; however, the signal transduction pathway that links reduced Ca(2+)-levels to RA synthesis remains unknown. Here we identify the Ca(2+)-dependent protein phosphatase calcineurin (CaN) as a key regulator for RA synthesis and homeostatic synaptic plasticity. Prolonged inhibition of CaN activity promotes RA synthesis in neurons, and leads to increased excitatory and decreased inhibitory synaptic transmission. These effects of CaN inhibitors on synaptic transmission are blocked by pharmacological inhibitors of RA synthesis or acute genetic deletion of the RA receptor RARα. Thus, CaN, acting upstream of RA, plays a critical role in gating RA signaling pathway in response to synaptic activity. Moreover, activity blockade-induced homeostatic synaptic plasticity is absent in CaN knockout neurons, demonstrating the essential role of CaN in RA-dependent homeostatic synaptic plasticity. Interestingly, in GluA1 S831A and S845A knockin mice, CaN inhibitor- and RA-induced regulation of synaptic transmission is intact, suggesting that phosphorylation of GluA1 C-terminal serine residues S831 and S845 is not required for CaN inhibitor- or RA-induced homeostatic synaptic plasticity. Thus, our study uncovers an unforeseen role of CaN in postsynaptic signaling, and defines CaN as the Ca(2+)-sensing signaling molecule that mediates RA-dependent homeostatic synaptic plasticity.
Keywords: AMPA receptor trafficking; calcineurin; homeostatic synaptic plasticity; retinoic acid; retinoic acid receptor RARα.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
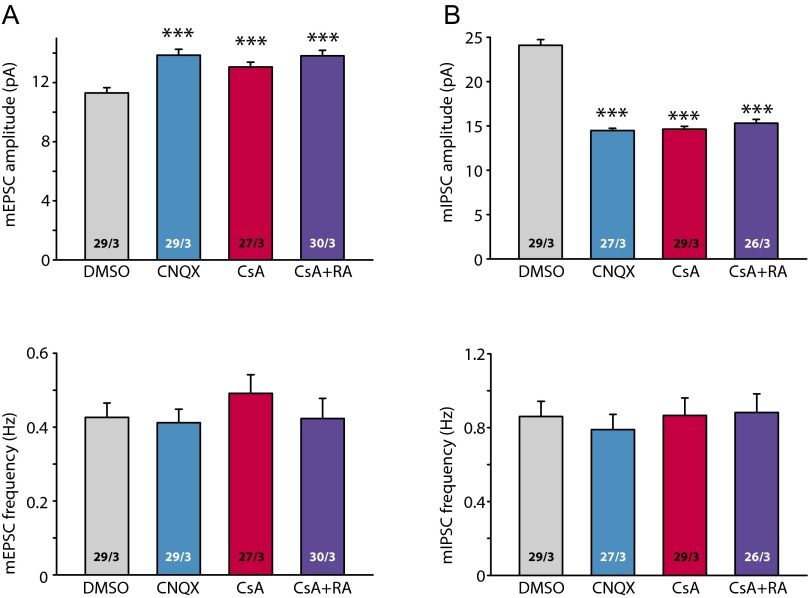
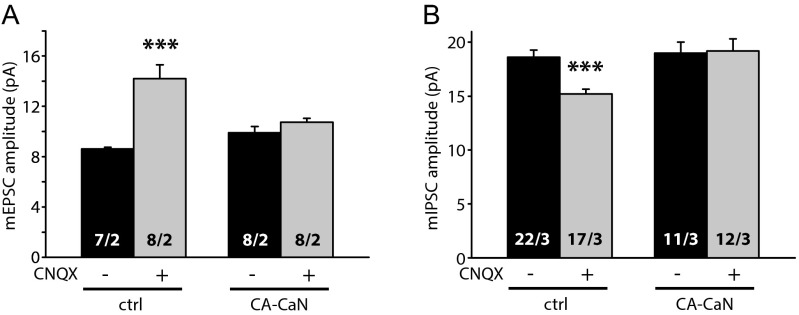
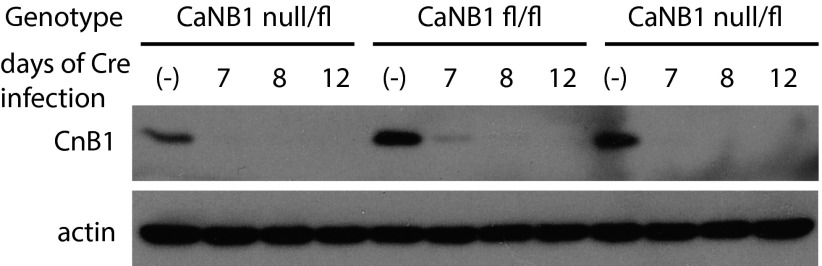
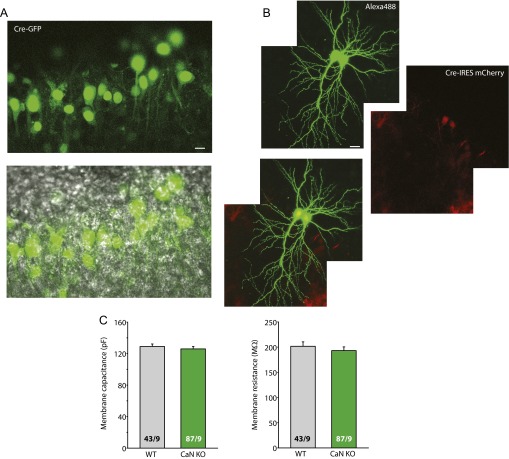
References
-
- Davis GW, Müller M. Homeostatic control of presynaptic neurotransmitter release. Annu Rev Physiol. 2015;77:251–270. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
