

Endoplasmic Reticulum Stress-Induced Autophagy Provides Cytoprotection from Chemical Hypoxia and Oxidant Injury and Ameliorates Renal Ischemia-Reperfusion Injury
- PMID: 26444017
- PMCID: PMC4596863
- DOI: 10.1371/journal.pone.0140025
Endoplasmic Reticulum Stress-Induced Autophagy Provides Cytoprotection from Chemical Hypoxia and Oxidant Injury and Ameliorates Renal Ischemia-Reperfusion Injury
Abstract
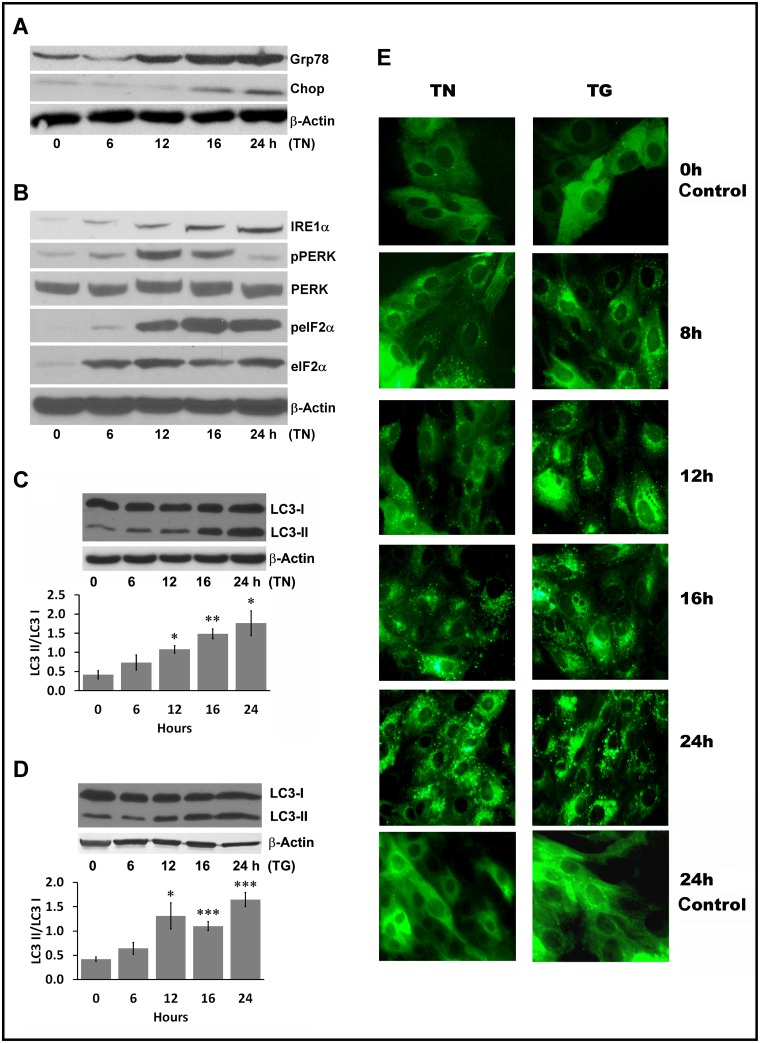
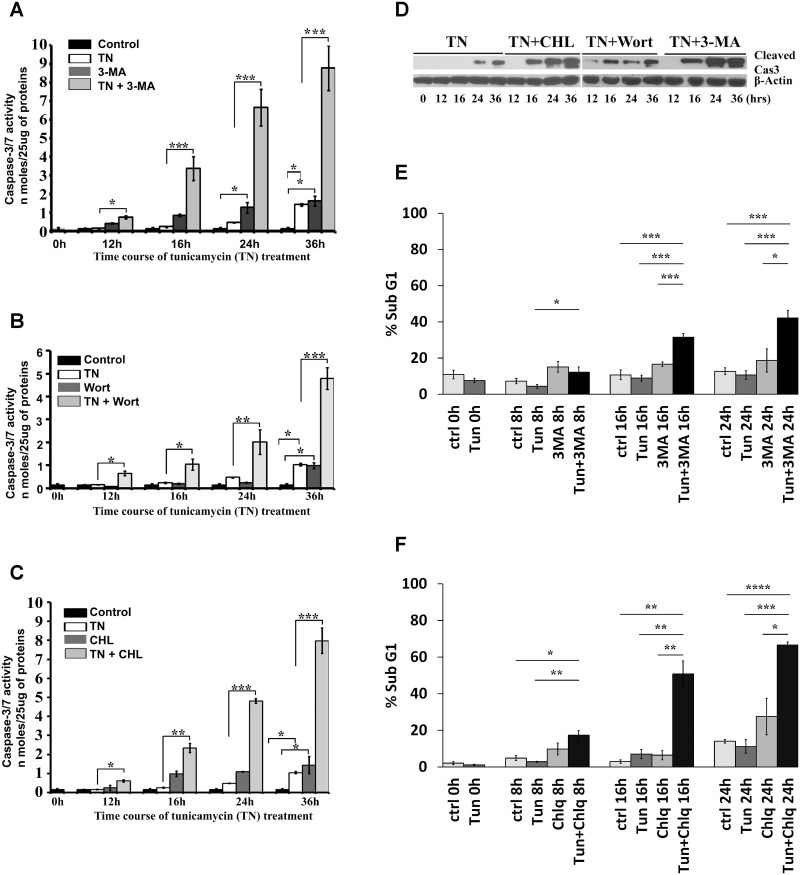
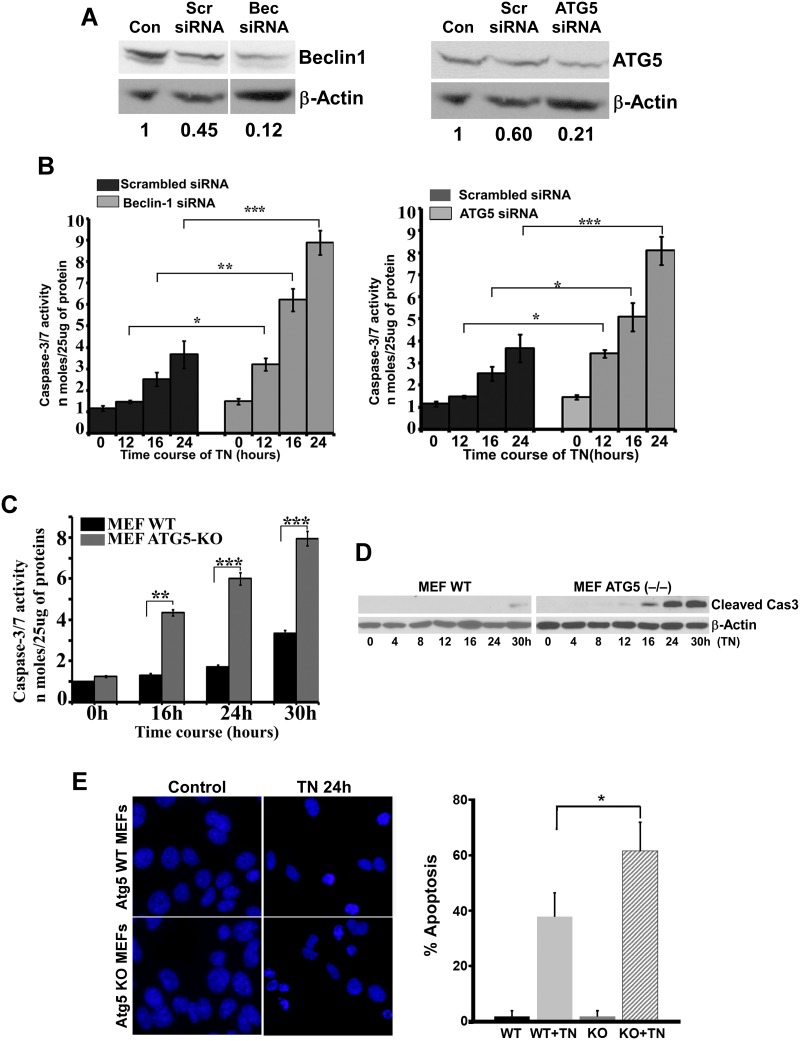
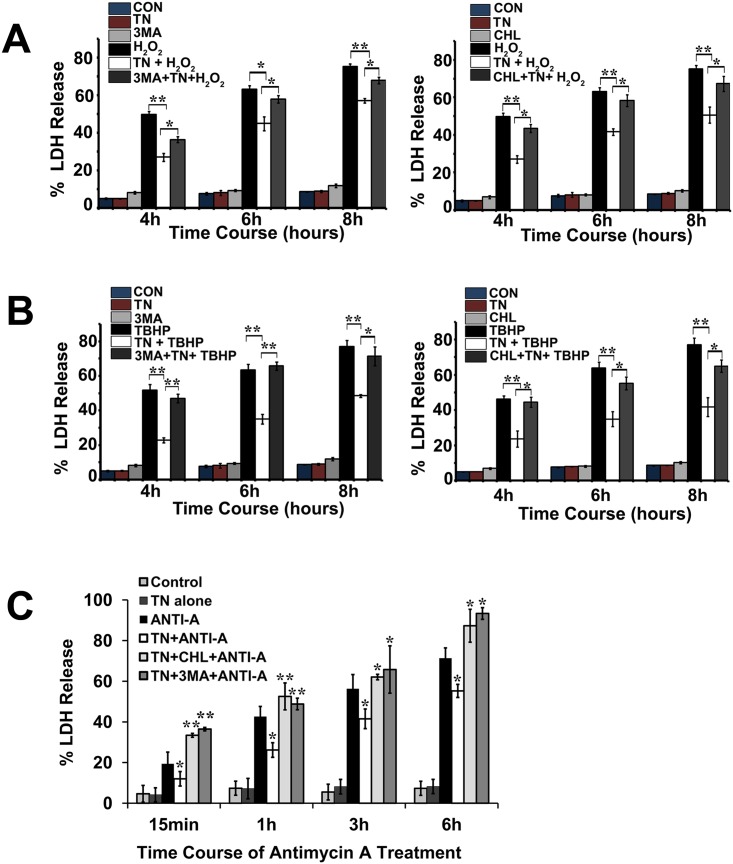
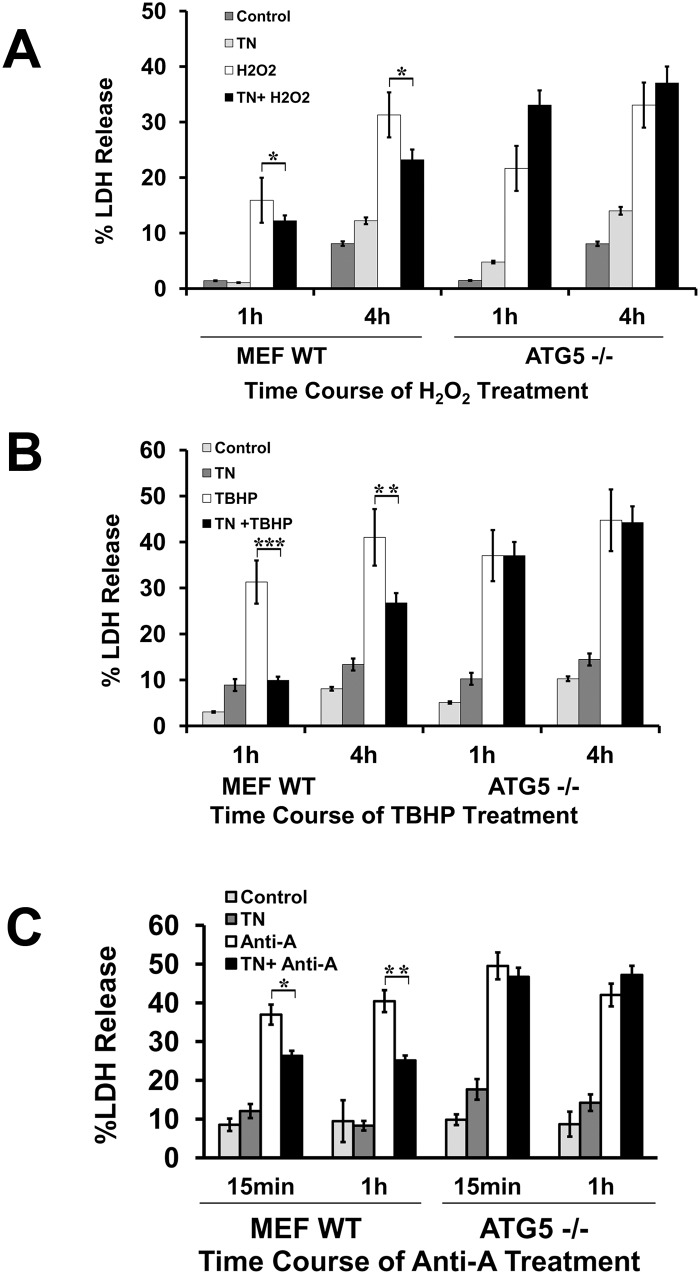
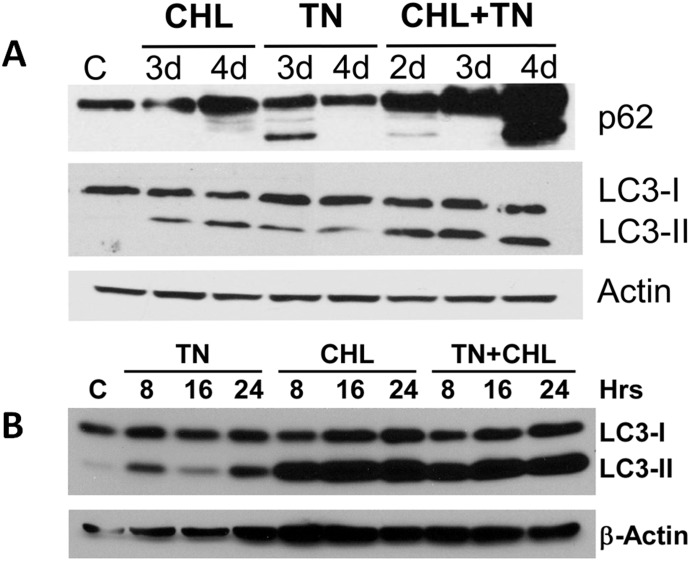
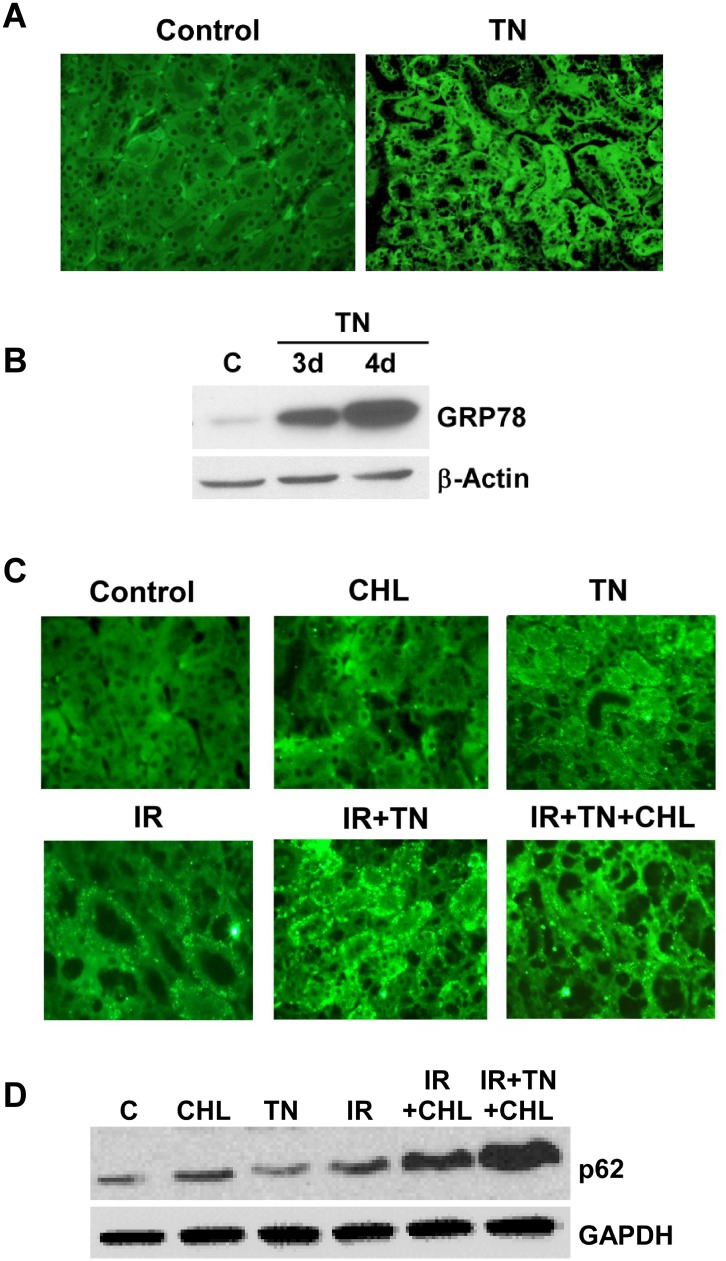
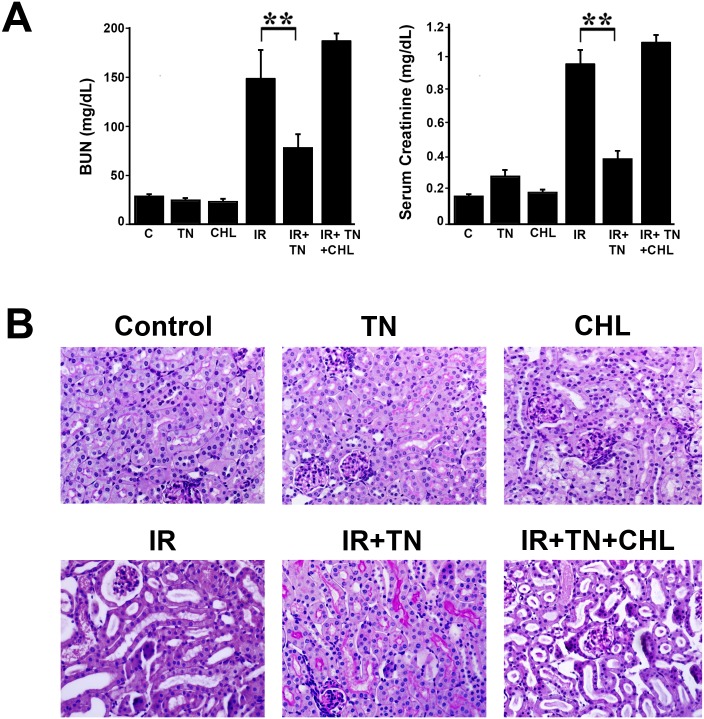
We examined whether endoplasmic reticulum (ER) stress-induced autophagy provides cytoprotection from renal tubular epithelial cell injury due to oxidants and chemical hypoxia in vitro, as well as from ischemia-reperfusion (IR) injury in vivo. We demonstrate that the ER stress inducer tunicamycin triggers an unfolded protein response, upregulates ER chaperone Grp78, and activates the autophagy pathway in renal tubular epithelial cells in culture. Inhibition of ER stress-induced autophagy accelerated caspase-3 activation and cell death suggesting a pro-survival role of ER stress-induced autophagy. Compared to wild-type cells, autophagy-deficient MEFs subjected to ER stress had enhanced caspase-3 activation and cell death, a finding that further supports the cytoprotective role of ER stress-induced autophagy. Induction of autophagy by ER stress markedly afforded cytoprotection from oxidants H2O2 and tert-Butyl hydroperoxide and from chemical hypoxia induced by antimycin A. In contrast, inhibition of ER stress-induced autophagy or autophagy-deficient cells markedly enhanced cell death in response to oxidant injury and chemical hypoxia. In mouse kidney, similarly to renal epithelial cells in culture, tunicamycin triggered ER stress, markedly upregulated Grp78, and activated autophagy without impairing the autophagic flux. In addition, ER stress-induced autophagy markedly ameliorated renal IR injury as evident from significant improvement in renal function and histology. Inhibition of autophagy by chloroquine markedly increased renal IR injury. These studies highlight beneficial impact of ER stress-induced autophagy in renal ischemia-reperfusion injury both in vitro and in vivo.
Conflict of interest statement
Figures
Similar articles
-
Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury.Am J Pathol. 2010 Mar;176(3):1181-92. doi: 10.2353/ajpath.2010.090594. Epub 2010 Jan 14. Am J Pathol. 2010. PMID: 20075199 Free PMC article.
-
Trehalose attenuates renal ischemia-reperfusion injury by enhancing autophagy and inhibiting oxidative stress and inflammation.Am J Physiol Renal Physiol. 2020 Apr 1;318(4):F994-F1005. doi: 10.1152/ajprenal.00568.2019. Epub 2020 Feb 18. Am J Physiol Renal Physiol. 2020. PMID: 32068461
-
Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury.Ann Med. 2018 Aug;50(5):381-390. doi: 10.1080/07853890.2018.1489142. Epub 2018 Jul 11. Ann Med. 2018. PMID: 29895209 Free PMC article. Review.
-
Roles of the Exogenous H2S-Mediated SR-A Signaling Pathway in Renal Ischemia/ Reperfusion Injury in Regulating Endoplasmic Reticulum Stress-Induced Autophagy in a Rat Model.Cell Physiol Biochem. 2017;41(6):2461-2474. doi: 10.1159/000475915. Epub 2017 May 4. Cell Physiol Biochem. 2017. Retraction in: Cell Physiol Biochem. 2021;55(5):663. doi: 10.33594/000000453. PMID: 28472786 Retracted.
-
Lipid droplets, autophagy, and ER stress as key (survival) pathways during ischemia-reperfusion of transplanted grafts.Cell Biol Int. 2024 Mar;48(3):253-279. doi: 10.1002/cbin.12114. Epub 2024 Jan 4. Cell Biol Int. 2024. PMID: 38178581 Review.
Cited by
-
Research progress on endoplasmic reticulum homeostasis in kidney diseases.Cell Death Dis. 2023 Jul 27;14(7):473. doi: 10.1038/s41419-023-05905-x. Cell Death Dis. 2023. PMID: 37500613 Free PMC article. Review.
-
Autophagy and Tubular Cell Death in the Kidney.Semin Nephrol. 2016 May;36(3):174-88. doi: 10.1016/j.semnephrol.2016.03.005. Semin Nephrol. 2016. PMID: 27339383 Free PMC article. Review.
-
Panobinostat mediated cell death: a novel therapeutic approach for osteosarcoma.Oncotarget. 2018 Aug 31;9(68):32997-33010. doi: 10.18632/oncotarget.26038. eCollection 2018 Aug 31. Oncotarget. 2018. PMID: 30250645 Free PMC article.
-
Renal tubular epithelial cells: the neglected mediator of tubulointerstitial fibrosis after injury.Cell Death Dis. 2018 Nov 13;9(11):1126. doi: 10.1038/s41419-018-1157-x. Cell Death Dis. 2018. PMID: 30425237 Free PMC article. Review.
-
A zebrafish HCT116 xenograft model to predict anandamide outcomes on colorectal cancer.Cell Death Dis. 2022 Dec 23;13(12):1069. doi: 10.1038/s41419-022-05523-z. Cell Death Dis. 2022. PMID: 36564370 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
